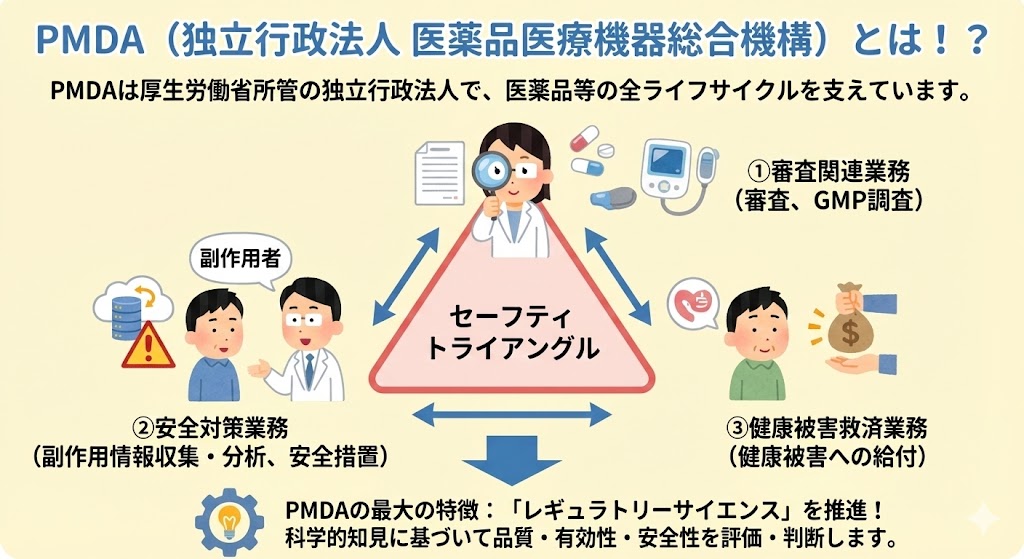

���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP�Ƃ́I�H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
GMP�ȗ߁i�����F�ߘa�R�N�W���P���{�s�j
�����\�Z�N�����J���ȗߑ�S���\�㍆
���i�y�ш�O�i�̐����Ǘ��y�ѕi���Ǘ��̊�Ɋւ���ȗ�
�@�@�i���a�O�\�ܔN�@����S�l�\�܍��j��\�l����l���y�ё�\����̓��܍��ɂ����ď��p�����\�l����l���̋K��Ɋ�Â��A���i�y�ш�O�i�̐����Ǘ��y�ѕi���Ǘ��K���i�����\��N�����ȗߑ�\�Z���j�̑S������������ȗ߂����̂悤�ɒ�߂�B
�ڎ�
���́@���i�����Ǝғ��̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ�
�@���߁@�ʑ��i��O���̎O�\���\���j
�@���߁@������i�̐����Ǘ��y�ѕi���Ǘ��i���\����\���\����j
�@��O�߁@���ۈ��i�̐����Ǘ��y�ѕi���Ǘ��i���\�O���\���\���j
�@��l�߁@�����R�����i���̐����Ǘ��y�ѕi���Ǘ��i���\���̓�\��O�\���j
�@��ܐ߁@�G���i��O�\����j
��O�́@��O�i�����Ǝғ��̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ�
�@���߁@�ʑ��i��O�\����\��l�\�����j
�@���߁@��O�i�̐����̗p�ɋ�����錴��̐����Ǘ��y�ѕi���Ǘ��i��l�\����E��\���j
�@��O�߁@���ۈ�O�i�̐����Ǘ��y�ѕi���Ǘ��i��\����\��\�O���j
���́@����
�i��|�j
�����@���̏ȗ߂́A���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���i���a�O�\�ܔN�@����S�l�\�܍��B�ȉ��u�@�v�Ƃ����B�j��\�l����l���i��\����̓��܍��ɂ����ď��p����ꍇ���܂ށB�ȉ������B�j�ɋK�肷������J���ȗ߂Œ�߂����߂���̂Ƃ���B
�i��`�j
�����@���̏ȗ߂Łu���i�v�Ƃ́A�������̐����H�����o�����i�����̒��ԍH���ő���ꂽ���̂ł����āA�Ȍ�̐����H�����o�邱�Ƃɂ���Đ��i�ƂȂ���́i�ȉ��u���Ԑ��i�v�Ƃ����B�j���܂ށB�j�������B
�Q�@���̏ȗ߂Łu�ŏI���i�v�Ƃ́A���i�̂����A���i�A��O�i�A���ϕi�y�эĐ���Ó����i�̕i���Ǘ��̊�Ɋւ���ȗ߁i�����\�Z�N�����J���ȗߑ�S�O�\�Z���j�����i���ߑ��\���ɂ����ď��p����ꍇ���܂ށB�j�̎s��ւ̏o�ׂ̉ۂ̌���ɋ��������̂������B
�R�@���̏ȗ߂Łu���ށv�Ƃ́A���i�̗e��A���y�ѕ\�����i�Y�t�������܂ށB�j�������B
�S�@���̏ȗ߂Łu���b�g�v�Ƃ́A��̐������ԓ��Ɉ�A�̐����H���ɂ��ώ�����L����悤�ɐ������ꂽ���i�y�ь����i�ȉ��u���i���v�Ƃ����B�j�̈�Q�������B
�T�@���̏ȗ߂Łu�Q�l�i�v�Ƃ́A�o�ׂ������i�ɕs����������ꍇ���A�o��ɐ��i�̕i�����Ċm�F����K�v���������ꍇ�ɔ����ĕۊǂ��鎎�������p�̌��̂������B
�U�@���̏ȗ߂Łu�ۑ��i�v�Ƃ́A�ŏI���i�̃��b�g����̎悳�ꂽ���̂ł����āA���ʂ��Ă��鐻�i�Ƃ̓��ꐫ���m�F���邽�߂Ɏg�p�������̂������B
�V�@���̏ȗ߂Łu���e�X�g���v�Ƃ́A�������ꂽ��������̊��Ԃ��o�߂������i���A���Y���Ԃ��o�߂������ȍ~�ɂ����āA������������̋K�i�ɓK�����Ă��邩�ǂ������ɂ��āA���߂Ď����������s���K�v��������̂Ƃ��Đݒ肳�����������B
�W�@���̏ȗ߂Łu�Ǘ��P�ʁv�Ƃ́A���ꐫ���m�F���ꂽ���ނ̈�Q�������B
�X�@���̏ȗ߂Łu���i�i���V�X�e���v�Ƃ́A���i�i�̊O�f�f�p���i�������B�ȉ������B�j�ɌW�鐻�i�̐����Ǝҋy�і@��\�O���̎O��ꍀ�ɋK�肷����i���O�������Ǝҁi�ȉ��u�O�������Ǝҁv�Ƃ����B�j�����Y���i�̕i���Ɋւ��ĊǗ��ē��s�����߂̃V�X�e���������B
�P�O�@���̏ȗ߂Łu�i�����X�N�}�l�W�����g�v�Ƃ́A���i�ɌW�鐻�i�ɂ��āA�i���ɍD�܂����Ȃ��e�����y�ڂ����ۋy�т��̔����m���i�ȉ��u�i�����X�N�v�Ƃ����B�j�̓���A�]���y�ъǗ������p���I�ɍs�����Ƃ������B
�P�P�@���̏ȗ߂Łu���萫���j�^�����O�v�Ƃ́A��߂�ꂽ�ۊǏ����̉��ŁA���i���L�����ԎႵ���͎g�p�̊����i�ȉ��P�Ɂu�L�����ԁv�Ƃ����B�j���̓��e�X�g���܂ł̊��Ԃɂ킽���ċK�i�ɓK�����Ă��邩�ǂ����ɂ��āA�p���I�Ɋm�F���邱�Ƃ������B
�P�Q�@���̏ȗ߂Łu�ƍ��v�Ƃ́A�ݒ肳�ꂽ�ڕW��B�������ł̑Ó����y�їL�����肷�邱�Ƃ������B
�P�R�@���̏ȗ߂Łu�o���f�[�V�����v�Ƃ́A�������̍\���ݔ����тɎ菇�A�H�����̑��̐����Ǘ��y�ѕi���Ǘ��̕��@�i�ȉ��u�����菇���v�Ƃ����B�j�����҂���錋�ʂ�^���邱�Ƃ������A������Ƃ��邱�Ƃ������B
�P�S�@���̏ȗ߂Łu�����[�u�v�Ƃ́A���m���ꂽ�s�K���i���̏ȗ߂ɋK�肷��v���������ɓK�����Ȃ����Ƃ������B�ȉ������B�j���̑��̖]�܂����Ȃ��̍Ĕ���h�~���邽�߁A���̌����ƂȂ�����Ԃ���������[�u�������B
�P�T�@���̏ȗ߂Łu�\�h�[�u�v�Ƃ́A��������s�K�����̑��̖]�܂����Ȃ��̔����𖢑R�ɖh�~���邽�߁A���̌����ƂȂ蓾���Ԃ���������[�u�������B
�P�U�@���̏ȗ߂Łu��ƊǗ����v�Ƃ́A���i���͈�O�i�ɌW�鐻�i�̐�����Ƃ��s���ꏊ�i�ȉ��u��Ə��v�Ƃ����B�j�̂����A��Ǝ��A�L��������\������Ă��āA�S�̂������x�ɐ���̈ێ����ł���悤�ɊǗ������ꏊ�������B
�P�V�@���̏ȗ߂Łu������v�Ƃ́A��Ə��̂����A�����̔��ʍ�Ƃ��s���ꏊ�A��܂̒�����Ƃ��s���ꏊ�y�ѐ���̗e�킪��Ə����̋�C�ɐG���ꏊ�������B
�P�W�@���̏ȗ߂Łu���ۋ��v�Ƃ́A��Ə��̂����A���ۉ����ꂽ��ܖ��͖ŋۂ��ꂽ�e�킪��Ə����̋�C�ɐG���ꏊ�A��܂̏[�U��Ƃ��s���ꏊ�A�e��̕Ǎ�Ƃ��s���ꏊ�y�і��ێ������̖��ۑ�����s���ꏊ�������B
�P�X�@���̏ȗ߂Łu�זE�g�D���i�v�Ƃ́A�l���͓����̍זE���͑g�D����\�����ꂽ���i�i�l�̌��t�y�ѐl�̌��t���琻������鐬������\���������i�������B�j�������B
�Q�O�@���̏ȗ߂Łu�����R�������v�Ƃ́A�@�����\���ɋK�肷�鐶���R�����i������i�i�ȉ��u�����R�����i�v�Ƃ����B�j�ɌW�鐻�i�̐����Ɏg�p���鐶���i�A���������B�j�ɗR�����錴���������B
�Q�P�@���̏ȗ߂Łu�h�i�[�v�Ƃ́A�זE�g�D���i�̌����ƂȂ�זE���͑g�D�����l�i����̈ڐA�Ɋւ���@���i������N�@����S�l���j��Z���ɋK�肷��]�������҂̐g�̂ɌW����̂������B�j�������B
�Q�Q�@���̏ȗ߂Łu�h�i�[�X�N���[�j���O�v�Ƃ́A�h�i�[�ɂ��āA��f�A�������ɂ��f�f���s���A�זE�g�D���i�ɌW�鐻�i�̌����ƂȂ�זE���͑g�D�����ɂ��\���ȓK�i����L���邩�ǂ����肷�邱�Ƃ������B
�Q�R�@���̏ȗ߂Łu�h�i�[�����v�Ƃ́A�זE�g�D���i�̌����ƂȂ�זE���͑g�D����铮���������B
�Q�S�@���̏ȗ߂Łu�h�i�[�����X�N���[�j���O�v�Ƃ́A�h�i�[�����ɂ��āA���������y�ю���Ǘ����s���A�זE�g�D���i�ɌW�鐻�i�̌����ƂȂ�זE���͑g�D�����ɂ��\���ȓK�i����L���邩�ǂ����肷�邱�Ƃ������B
�i�K�p�͈̔́j
��O���@�@��\�l���ꍀ�ɋK�肷����i���͈�O�i�̐����̔��Ǝҁi�@��\����̓��l���ɋK�肷��I�C�O���������i�������̔��Ǝ҂��܂ށB�ȉ������B�j�́A���i�ɂ����Ă͑��́A��O�i�ɂ����Ă͑�O�͂̋K��Ɋ�Â��A���i���͈�O�i�ɌW�鐻�i�̐����Ǝҋy�ъO�������Ǝҁi�ȉ��u�����Ǝғ��v�Ƒ��̂���B�j�ɐ������ɂ����鐻���Ǘ��y�ѕi���Ǘ����s�킹�Ȃ���Ȃ�Ȃ��B
�Q�@���i���͈�O�i�ɌW�鐻�i�̐����Ǝғ��́A���i�ɂ����Ă͑��́A��O�i�ɂ����Ă͑�O�͂̋K��Ɋ�Â��A���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���{�s�K���i���a�O�\�Z�N�����ȗߑ�ꍆ�B�ȉ��u�{�s�K���v�Ƃ����B�j���\�Z���ɋK�肷�鐻�����ɂ����鐻�i�̐����Ǘ��y�ѕi���Ǘ����s��Ȃ���Ȃ�Ȃ��B
�R�@�@�攪�\���ꍀ�ɋK�肷��A�o�p�̈��i���͈�O�i�ɌW�鐻�i�̐����Ǝ҂́A���i�ɂ����Ă͑��́A��O�i�ɂ����Ă͑�O�͂̋K��Ɋ�Â��A���Y���i�̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ����s��Ȃ���Ȃ�Ȃ��B
�i���F�����̏���j
��O���̓�@�@��\�l���ꍀ�ɋK�肷����i���͈�O�i�ɌW�鐻�i�̐����Ǝғ��́A���Y���i��@��\�l���ꍀ�Ⴕ���͓����\�܍��i�@��\����̓��܍��ɂ����ď��p����ꍇ���܂ށB�ȉ����̏��ɂ����ē����B�j���͖@��\����̓��ꍀ�̏��F���������i�ȉ��u���F�����v�Ƃ����B�j�ɏ]���Đ������Ȃ���Ȃ�Ȃ��B�������A�@��\�l���\�܍��̌y���ȕύX���s���ꍇ�ɂ����ẮA�����\�Z���i�@��\����̓��܍��ɂ����ď��p����ꍇ���܂ށB�j�̋K��ɂ��͏o���s����܂ł̊Ԃ́A���̌���łȂ��B
���́@���i�����Ǝғ��̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ�
���߁@�ʑ�
�i���i�i���V�X�e���j
��O���̎O�@�����Ǝғ��́A�������̂�����i�i���V�X�e�����\�z����ƂƂ��ɁA���Ɍf����Ɩ����s��Ȃ���Ȃ�Ȃ��B
��@���i�i�����m�ۂ��邽�߂̊�{�I�ȕ��j�i�ȉ��u�i�����j�v�Ƃ����B�j���ɂ���߁A���Y�����Ɉ��i�i���V�X�e���̎葱���̍\���v�f���������ƁB
��@�@��\�����Z���ɋK�肷����i�����Ǘ��ҋy�і@��Z�\�����̏\�Z��ꍀ�ɋK�肷�鐶���R�����i�̐������Ǘ�����ҁi�O�������Ǝ҂ɂ����ẮA�@��\�O���̎O��ꍀ�̔F������������̐ӔC�Җ��͓��Y�O�������Ǝ҂����炩���ߎw�肵���ҁj�i�ȉ��u�����Ǘ��ҁv�Ƒ��̂���B�j���͑�l���O����ꍆ�ɋK�肷��i���ۏɌW��Ɩ���S������g�D�ɁA�i�����j�Ɋ�Â����������ɂ�����i���ڕW���A�����ɂ���߂����邱�ƁB
�O�@�������ɂ����Ĉ��i�i���V�X�e���Ɋւ��S�Ă̑g�D�y�ѐE���ɑ��A�i�����j�y�ѕi���ڕW�����m���邱�ƁB
�l�@�i�����j�y�ѕi���ڕW��B�����邽�߁A�K�v�Ȏ����i�l�̗L����m���y�ыZ�\���тɋZ�p�A�ݔ����̑��̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ��Ɋ��p����鎑���������B�j��z������ƂƂ��ɁA����I�Ɉ��i�i���V�X�e�����ƍ����A���̌��ʂɊ�Â��ď��v�̑[�u���u���邱�ƁB
�܁@�O�̋Ɩ��ɌW��L�^���A���炩���ߎw�肵���҂ɍ쐬�����A�����ۊǂ����邱�ƁB
�i�i�����X�N�}�l�W�����g�j
��O���̎l�@�����Ǝғ��́A�i�����X�N�}�l�W�����g�����p���Ĉ��i�i���V�X�e�����\�z������ŁA���i�ɌW�鐻�i�ɂ��āA�������ɂ����鐻���Ǘ��y�ѕi���Ǘ����s��Ȃ���Ȃ�Ȃ��B
�Q�@�����Ǝғ��́A���炩���ߎw�肵���҂ɕi�����X�N�}�l�W�����g�̎��{�̎葱���̑��̕K�v�Ȏ����ɌW�镶���y�ыL�^���쐬�����A������ۊǂ����Ȃ���Ȃ�Ȃ��B
�i��������y�ѕi������j
��l���@�����Ǝғ��́A���������ƂɁA�����Ǘ��҂̊ē̉��ɁA�����Ǘ��ɌW�镔��i�ȉ��u��������v�Ƃ����B�j�y�ѕi���Ǘ��ɌW�镔��i�ȉ��u�i������v�Ƃ����B�j��u���Ȃ���Ȃ�Ȃ��B
�Q�@�i������́A�������傩��Ɨ����Ă��Ȃ���Ȃ�Ȃ��B
�R�@�i������́A���Ɍf����g�D��u���Ȃ���Ȃ�Ȃ��B
��@�i���ۏɌW��Ɩ���S������g�D
��@���������i�����Ǝғ��̑��̎��������ݔ��𗘗p�����͑�\����̌܂̋K��ɏ]���đ��Ɉϑ����Ď��Ȃ̐ӔC�ɂ����čs�����������ł����āA���Y���p���͈ϑ��ɂ��x�Ⴊ�Ȃ��ƔF�߂�����̂��܂ށB�ȉ����̏͂ɂ����ē����B�j�ɌW��Ɩ���S������g�D
�i�����Ǘ��ҁj
����@�����Ǘ��҂́A���Ɍf����Ɩ����s��Ȃ���Ȃ�Ȃ��B
��@�i�����j�y�ѕi���ڕW��B�����邽�߁A�������ɂ����āA�����Ǘ��A�i���ۏ؋y�ю��������ɌW��Ɩ��i�ȉ��u�����E�i���֘A�Ɩ��v�Ƃ����B�j���K�����~���ɍs����悤��������ƂƂ��ɁA���i�i���V�X�e�����K�ɉ^�p�����悤�Ǘ����邱�ƁB
��@���i�i���V�X�e���̉^�p���m�F����ƂƂ��ɁA���̉��P��v���邩�ǂ����ɂ��Đ����Ǝғ��ɑ��ĕ����ɂ����邱�ƁB
�O�@�����A���ދy�ѐ��i�̋K�i���тɐ����菇�������F�����Ƒ��Ⴗ�邱�Ƃ̂Ȃ��悤�A�i���ۏɌW��Ɩ���S������g�D�ɊǗ������邱�ƁB
�l�@�i���s�ǂ��̑����i�i���ɏd��ȉe�����y�Ԃ����ꂪ����ꍇ�ɂ����ẮA���v�̑[�u�����₩�ɂƂ��Ă��邱�Ƌy�т��̐i�����悭���m�F���A�K�v�ɉ����A���P�����v�̑[�u���Ƃ�悤�w�����邱�ƁB
�Q�@�����Ǝғ��́A�����Ǘ��҂��Ɩ����s���ɓ������Ďx����邱�Ƃ��Ȃ��悤�ɂ��Ȃ���Ȃ�Ȃ��B
�i�E���j
��Z���@�����Ǝғ��́A�����E�i���֘A�Ɩ���K�����~���Ɏ��{������\�͂�L����ӔC�ҁi�ȉ����̏͂ɂ����ĒP�Ɂu�ӔC�ҁv�Ƃ����B�j���A�������̑g�D�A�K�͋y�ыƖ��̎�ޓ��ɉ����A�K�ɒu���Ȃ���Ȃ�Ȃ��B
�Q�@�����Ǝғ��́A�������̑g�D�A�K�͋y�ыƖ��̎�ޓ��ɉ����A�K�Ȑl���̐ӔC�҂�z�u���Ȃ���Ȃ�Ȃ��B
�R�@�����Ǝғ��́A�����E�i���֘A�Ɩ���K�Ɏ��{������\�͂�L����l�����\���Ɋm�ۂ��Ȃ���Ȃ�Ȃ��B
�S�@�����Ǝғ��́A�����E�i���֘A�Ɩ��ɏ]������E���i�����Ǘ��ҋy�ѐӔC�҂��܂ށB�j�̐Ӗ��y�ъǗ��̐����ɂ��K�ɒ�߂Ȃ���Ȃ�Ȃ��B
�i���i���i�W�����j
�掵���@�����Ǝғ��́A���i�ɌW�鐻�i�i���Ԑ��i�������B�j�Ɋւ��Ď��Ɍf���鎖���ɂ��ċL�ڂ��������i�ȉ��u���i���i�W�����v�Ƃ����B�j�Y���i�̐����ɌW�鐻�������Ƃɍ쐬���A�i������̏��F���A���Y�������ɓK�ɔ����u���Ȃ���Ȃ�Ȃ��B
��@���F�����̂����A���Y�������ɂ����鐻�����@�A�K�i�y�ю������@���̑��̕K�v�Ȏ���
��@�@��l�\����ꍀ�̋K��ɂ���߂�ꂽ����̑��Ɋւ���@�ߖ��͂���Ɋ�Â����ߎႵ���͏����̂����i���Ɋւ��鎖��
�O�@�����菇�i��ꍆ�̎����������B�j
�l�@���̑����v�̎���
�i�菇�����j
�攪���@�����Ǝғ��́A���������ƂɁA���Ɍf����菇�ɂ��ċL�ڂ��������i�ȉ��u�菇���v�Ƃ����B�j���쐬���A����Y�������ɓK�ɔ����u���Ȃ���Ȃ�Ȃ��B
��@�\���ݔ��y�ѐE���̉q���Ǘ��Ɋւ���菇
��@�����H���A�����ݔ��A�����A���ދy�ѐ��i�̊Ǘ��Ɋւ���菇
�O�@���������ݔ��y�ь��̂̊Ǘ����̑��K�Ȏ��������̎��{�ɕK�v�Ȏ菇
�l�@���萫���j�^�����O�Ɋւ���菇
�܁@���i�i���̏ƍ��Ɋւ���菇
�Z�@�����y�ю��ށi�ȉ��u�������v�Ƃ����B�j�̋����҂̊Ǘ��Ɋւ���菇
���@�����Ǝғ��̈ϑ����Ď����������̑��̐����E�i���֘A�Ɩ��̈ꕔ���s�����̎��Ǝҁi�ȉ��u�O���ϑ��Ǝҁv�Ƃ����B�j�̊Ǘ��Ɋւ���菇
���@����������̏o�ׂ̊Ǘ��Ɋւ���菇
��@�o���f�[�V�����Ɋւ���菇
�\�@��\�l���̕ύX�̊Ǘ��Ɋւ���菇
�\��@��\���̈�E�̊Ǘ��Ɋւ���菇
�\��@��\�Z���̕i�����y�ѕi���s�Ǔ��̏����Ɋւ���菇
�\�O�@������̏����Ɋւ���菇
�\�l�@���ȓ_���Ɋւ���菇
�\�܁@����P���Ɋւ���菇
�\�Z�@�����y�ыL�^�̍쐬�A�����y�ѕۊǂɊւ���菇
�\���@���̑��K�����~���Ȑ����E�i���֘A�Ɩ��ɕK�v�Ȏ菇
�Q�@�����Ǝғ��́A���i���i�W�����y�ю菇���i�ȉ����̏͂ɂ����āu�菇�����v�Ƒ��̂���B�j���тɂ��̏͂ɋK�肷��L�^�ɂ��āA���̐M�������p���I�Ɋm�ۂ��邽�߁A���\���e���Ɍf����Ɩ��̕��@�Ɋւ��鎖�����A�����ɂ���߂Ȃ���Ȃ�Ȃ��B
�i���������̖h�~�j
�攪���̓�@�����Ǝғ��́A���i�ɌW�鐻�i�̌���������h�~���邽�߁A�����菇���ɂ��ď��v�̑[�u���Ƃ�Ȃ���Ȃ�Ȃ��B
�i�\���ݔ��j
�����@���i�ɌW�鐻�i�̐������̍\���ݔ��́A���ɒ�߂�Ƃ���ɓK��������̂łȂ���Ȃ�Ȃ��B
��@�菇�����Ɋ�Â��A���̗p�r�ɉ����K�ɐ��|�y�ѕێ炪�s���A�K�v�ɉ����ŋۂ���A�܂��A���̋L�^���쐬����A�ۊǂ���Ă��邱�ƁB
��@���i���ɂ��L�ŃK�X����舵���ꍇ�ɂ����ẮA���̏����ɗv����ݔ���L���邱�ƁB
�O�@��Ə��̂�����Ǝ��́A���i�̎�ށA�܌`�y�ѐ����H���ɉ����A�������͔������ɂ�鉘����h�~����̂ɕK�v�ȍ\���y�ѐݔ���L���Ă��邱�ƁB�������A�����ݔ����̗L����@�\�ɂ�肱��Ɠ����x�̌��ʂ���ꍇ�ɂ����ẮA���̌���łȂ��B
�l�@��Ə��̂����A�����̔��ʍ�ƁA���i�̒�����ƁA�[�U��Ɩ��͕Ǎ�Ƃ��s����Ǝ��́A���Y��Ǝ��̐E���ȊO�̎҂̒ʘH�ƂȂ�Ȃ��悤�ɑ����Ă��邱�ƁB�������A���Y��Ǝ��̐E���ȊO�̎҂ɂ�鐻�i�ւ̉����̂����ꂪ�Ȃ��ꍇ�ɂ����ẮA���̌���łȂ��B
�܁@���Ɍf����ꍇ�ɂ����ẮA���i������舵����Ǝ��i���e��Ɏ��߂�ꂽ���i���݂̂���舵����Ǝ��y�ѐ��i������̎悳�ꂽ���݂̂̂���舵����Ǝ��������B�����ɂ����ē����B�j���p�Ƃ��A���A��C�����V�X�e����ʌn���ɂ��铙�̓��Y���i���̘R�o��h�~����K�ȑ[�u���Ƃ��Ă��邱�ƁB
�C�@��U���₷���A���ʂʼnߕq�ǔ������������i������舵���ꍇ
���@�����������邱�Ƃɂ�葼�̐��i���ɏd��ȉe�����y�Ԃ�����̂��鐻�i���i������p���͓Ő���L������̂��܂ށB�j����舵���ꍇ�ł����āA����������h�~����K�ȑ[�u���Ƃ邱�Ƃ��ł��Ȃ��ꍇ
�Z�@���i�̐����ɕK�v�Ȏ��y�їʂ̐��i�ݔ��y�ъ����тɗe��̐���܂ށB�j����������ݔ���L���邱�ƁB
�Q�@���i������舵����Ǝ��ŁA���̏ȗ߂��K�p����Ȃ����i�̐�����Ƃ��s���Ă͂Ȃ�Ȃ��B�������A���炩���ߌ����ꂽ�H�����͐��ɂ���ē��Y���i�̐�����K�ɕs�������͏������A���i�ɌW�鐻�i�Ƃ̌���������h�~����K�ȑ[�u���Ƃ�ꍇ�i���Ɍf����ꍇ�������B�j�ɂ����ẮA���̌���łȂ��B
��@���Y���i�̐�����Ƃɂ����āA��U���₷���A���ʂʼnߕq�ǔ�����������������舵���ꍇ
��@���Y���i���l�̐g�̂Ɏg�p����邱�Ƃ��ړI�Ƃ���Ă��Ȃ����̂ł����āA���A���̐�����������p�y�ѓŐ���L���Ȃ����Ƃ����炩�łȂ��ꍇ
�i�����Ǘ��j
��\���@�����Ǝғ��́A��������ɁA�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�����H���ɂ�����w�������A���ӎ������̑��K�v�Ȏ������L�ڂ��������i�ȉ��u�����w�}���v�Ƃ����B�j���쐬���A�����ۊǂ��邱�ƁB
��@��������̐ӔC�҂��A�����w�}���Ɋ�Â��A���i�̐�����Ƃɏ]������E���ɑ��ē��Y��Ƃ��w�����邱�ƁB
�O�@�����w�}���Ɋ�Â��A���i�̐�����Ƃ��s�����ƁB�܂��A���b�g���\�����鐻�i�ɂ��ẮA�����Ƃ��āA��̐����w�}���Ɋ�Â��Đ������ꂽ���i�̈�Q����̃��b�g�ƂȂ�悤������Ƃ��s�����ƁB
�l�@�����Ɋւ���L�^�����b�g���Ɓi���b�g���\�����Ȃ����i���ɂ��Ă͐����ԍ����ƁB���\�����ꍀ�������A�ȉ������B�j�ɍ쐬���A�����ۊǂ��邱�ƁB
�܁@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɁA���ꂪ�K���ł���|���m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�Z�@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɓK���ɕۊǂ��A�o�[���s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
���@�\���ݔ��̐�����m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
���@�E���̉q���Ǘ����s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@�\���ݔ������I�ɓ_����������ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB�܂��A�v��̍Z����K�ɍs���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�\�@�����A�ۊNjy�яo�[���тɉq���Ǘ��Ɋւ���L�^�ɂ�萻���Ǘ����K�ɍs���Ă��邱�Ƃ��m�F���A���̌��ʂ�i���ۏɌW��Ɩ���S������g�D�ɑ��ĕ����ɂ����邱�ƁB
�\��@���̑������Ǘ��̂��߂ɕK�v�ȋƖ�
�i�i���Ǘ��j
��\����@�����Ǝғ��́A�i������ɁA�菇�����Ɋ�Â��A���Ɍf����i���ۏ؋y�ю��������ɌW��Ɩ����v��I���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɎ����������s���̂ɕK�v�Ȍ��̂��̎悷��ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@�̎悵�����̋y�т��̎��������p�̕W���i��K�ɕۊǂ��邱�ƁB
�O�@�i������̐ӔC�҂��A�����A���ދy�ѐ��i�̎��������ɏ]������E���ɑ��āA���Y��Ƃɂ������ɂ��w�����邱�ƁB
�l�@�̎悵�����̂ɂ��āA�O���̕����Ɋ�Â��A���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɎ����������s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�܁@�ŏI���i�i���b�g���\��������̂Ɍ���B�j�ɂ��āA���b�g���Ƃɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��Q�l�i�Ƃ��āA�������ꂽ�����瓖�Y���i�̗L�����ԂɈ�N�i���ː����i�̍ŏI���i�ɂ����ẮA�Z�����͕i�����X�N�}�l�W�����g�Ɋ�Â��K�ȓ����j�����Z�������ԓK�ȕۊǏ����̉��ŕۊǂ��邱�ƁB�܂��A���̕ۑ��i�Y�Q�l�i�Ɠ����ԕۊǂ��邱�ƁB
�Z�@���i�ɌW�鐻�i�̐����Ɏg�p�����������̂������Y���i�̕i���ɉe�����y�ڂ����̂ɂ��āA�����ɂ����Ă̓��b�g���Ƃɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��A���ނɂ����Ă͊Ǘ��P�ʂ��Ƃɏ���̎��������ɕK�v�ȗʂ��A���ꂼ��Q�l�i�Ƃ��āA���Y���i�̏o�ׂ肵���������N�ԁi���ː����i�ɌW�鐻�i�̌����ɂ����ẮA���Y�����̈��萫�Ɋ�Â��K�Ȋ��ԁj�K�ȕۊǏ����̉��ŕۊǂ��邱�ƁB
���@���������Ɋւ���ݔ��y�ъ������I�ɓ_����������ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB�܂��A���������Ɋւ���v��̍Z����K�ɍs���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
���@��l���̎��������̌��ʂ̔�����s���A���̌��ʂ�����ɑ��ĕ����ɂ����邱�ƁB�܂��A���Y���������ɂ��āA�K�i�ɓK�����Ȃ����ʂƂȂ����ꍇ�ɂ����ẮA���̌������������A���v�̐����[�u�y�ї\�h�[�u���Ƃ�ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@���̑��i���ۏ؋y�ю��������̂��߂ɕK�v�ȋƖ�
�Q�@�A���捑�ɂ����鐻���Ǘ��y�ѕi���Ǘ��̊���тɂ����̊�ɑ���K�����̊m�F�Ɋւ���葱���䂪���̂��̂Ɠ����ł���ƔF�߂���ꍇ�ɂ����ẮA�����Ǝ҂́A�A�����i�ɌW��O����l���ɋK�肷�鎎�������i�O�ό����������B�j���A���Y�A�����i�ɂ��ėA���捑�̊O�������Ǝ҂��s�������������̋L�^���m�F���邱�Ƃ������đウ�邱�Ƃ��ł���B���̏ꍇ�ɂ����āA�����Ǝ҂́A�i���ۏɌW��Ɩ���S������g�D�ɁA���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�A�����i���A���Y�O�������Ǝ҂̐������ɂ����āA�K�Ȑ����菇���ɂ�萻������Ă��邱�Ƃ����I�Ɋm�F���邱�ƁB
��@���Y�O�������Ǝ҂̐��������A���̍��ɂ����鐻���Ǘ��y�ѕi���Ǘ��Ɋւ����ɓK�����Ă��邱�Ƃ����I�Ɋm�F���邱�ƁB
�O�@�O�̊m�F�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�l�@���Y�A�����i�ɂ��ē��Y�O�������Ǝ҂��s�������������̋L�^���m�F����ƂƂ��ɁA���̊m�F�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�R�@�����Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A�O���\���̋K��ɂ�萻�����傩����ꂽ�����Ǘ��ɌW��m�F�̌��ʂ����b�g���ƂɊm�F�����Ȃ���Ȃ�Ȃ��B
�i���萫���j�^�����O�j
��\����̓�@�ŏI���i������i�̐����Ǝғ��́A���Y���i�ɂ��āA�i������ɁA�菇�����Ɋ�Â��A���Ɍf������萫���j�^�����O�ɌW��Ɩ����v��I���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�i�����X�N����肵�A�]�����s�������ʂɊ�Â��āA���萫���j�^�����O���s�����i��K�ɑI�肵�A�K�v�ʂ̌��̂��̎悷�邱�ƁB
��@���Y���i�̋K�i�̂����ۑ��ɂ��e�����₷�����ڋy�ѓ��Y�K�i�ɓK�����Ȃ��ꍇ�ɓ��Y���i�̗L�������͈��S���ɉe�����y�ڂ��ƍl�����鍀�ڂ��A���������̍��ڂƂ��đI�肷�邱�ƁB
�O�@��ꍆ�̌��̂�ۊǂ��A�O���̍��ڂɂ��āA�K�ȊԊu�Ŏ����������s�����ƁB
�l�@�O���̎��������̌��ʂɊ�Â��A���Y���i�̕i���ւ̉e����]�����邱�ƁB
�܁@�O�e���̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�ŏI���i������i�̐����Ǝғ��́A�O����l���̕]���̌��ʂ���A���Y���i�̋K�i�ɓK�����Ȃ��ꍇ���͂��̂����ꂪ����ꍇ�ɂ����ẮA���Y���i�ɌW�鐻���̔��Ǝ҂ɑ��鑬�₩�ȘA���A���i�̉���̔��f�ɕK�v�ȏ��̒��A���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�ɌW��L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i���i�i���̏ƍ��j
��\����̎O�@�����Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�����H�����тɌ����A���ދy�ѐ��i�̋K�i�̑Ó����������邱�Ƃ�ړI�Ƃ��āA����I���͐����ɁA���i�i���̏ƍ����s�����ƁB
��@�O���̏ƍ��̌��ʂ��Ǘ��҂ɑ��ĕ����ɂ����邱�ƁB
�Q�@�����Ǝғ��́A�O����ꍆ�̏ƍ��̌��ʂɊ�Â��A�����Ǘ��Ⴕ���͕i���Ǘ��Ɋւ��ĉ��P��v����ꍇ���̓o���f�[�V�������s�����Ƃ�v����ꍇ�ɂ����ẮA���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i�������̋����҂̊Ǘ��j
��\����̎l�@�����Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�������̕i���̊m�ۂ̂��߂ɓK�ȋK�i���߂邱�ƁB
��@�������̋����҂ɂ��āA�K�i����]��������őI�肷�邱�ƁB
�O�@�������̐����Ǘ��y�ѕi���Ǘ����K���~���ɍs���Ă��邩�ǂ����ɂ��Ē���I�Ɋm�F���邱�ƁB
�l�@�O�O���̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�������̂������i�i���ɉe�����y�ڂ����̂ɂ��āA���Y�������̐����Ǘ��y�ѕi���Ǘ��̕��@�Ɋւ��Ă��̋����҂ƕ����ɂ��K�v�Ȏ挈�߂�������Ȃ���Ȃ�Ȃ��B�������A���Y�挈�߂��A���Y���������g�p���鐻�i�ɌW�鐻���̔��ƎҖ��͖@��\����̓��ꍀ�̏��F�����҂Ɠ��Y�����҂Ƃ̊Ԃɂ����Ē�������Ă���ꍇ�ɂ����ẮA���̌���łȂ��B
�i�O���ϑ��Ǝ҂̊Ǘ��j
��\����̌܁@�����Ǝғ��́A�����������̑��̐����E�i���֘A�Ɩ��̈ꕔ�i���̎��Ǝ҂ɍs�킹�邱�Ƃɂ��x�Ⴊ�Ȃ��ƔF�߂�����̂Ɍ���B�j���O���ϑ��Ǝ҂Ɉϑ�����ꍇ�ɂ����ẮA���Y�O���ϑ��Ǝ҂ƕ����ɂ��K�v�Ȏ挈�߂�������Ȃ���Ȃ�Ȃ��B�������A���Y�挈�߂��A���Y�����E�i���֘A�Ɩ����s���鐻�i�ɌW�鐻���̔��ƎҖ��͖@��\����̓��ꍀ�̏��F�����҂Ɠ��Y�O���ϑ��Ǝ҂Ƃ̊Ԃɂ����Ē�������Ă���ꍇ�ɂ����ẮA���̌���łȂ��i������ꍆ�ɂ����ē����B�j�B
�Q�@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�O���ϑ��Ǝ҂Ƃ̎挈�߂̒����ɍۂ��āA���Y�O���ϑ��Ǝ҂̓K���y�є\�͂ɂ��Ċm�F���邱�ƁB
��@�O���ϑ��Ǝ҂����Y�ϑ��ɌW�鐻���E�i���֘A�Ɩ���K�����~���ɍs���Ă��邩�ǂ����ɂ��Ē���I�Ɋm�F����ƂƂ��ɁA�K�v�ɉ����ĉ��P�����߂邱�ƁB
�O�@�O�̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�i����������̏o�ׂ̊Ǘ��j
��\����@�����Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A�����E�i���֘A�Ɩ����K�ɍs��ꂽ���ǂ����ɂ��ă��b�g���ƂɓK�ɕ]�����A���i�̐���������̏o�ׂ̉ۂ����肷��Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
�Q�@�O���̋Ɩ����s���҂́A���Y�Ɩ���K�����~���Ɏ��{������\�͂�L����҂łȂ���Ȃ�Ȃ��B
�R�@�����Ǝғ��́A��ꍀ�̋Ɩ����s���҂����Y�Ɩ����s���ɓ������āA�x�Ⴊ�����邱�Ƃ��Ȃ��悤�ɂ��Ȃ���Ȃ�Ȃ��B
�S�@�����Ǝғ��́A��ꍀ�̌��肪�K���ɍs����܂Ő��������琻�i���o�ׂ��Ă͂Ȃ�Ȃ��B
�i�o���f�[�V�����j
��\�O���@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Ɍf����ꍇ�ɂ����ăo���f�[�V�������s�����ƁB
�C�@���Y�������ɂ����ĐV���Ɉ��i�̐������J�n����ꍇ
���@�����菇���ɂ��Đ��i�i���ɑ傫�ȉe�����y�ڂ��ύX������ꍇ
�n�@���̑����i�̐����Ǘ��y�ѕi���Ǘ���K�ɍs�����߂ɕK�v�ƔF�߂���ꍇ
��@�o���f�[�V�����̌v��y�ь��ʂ��A�i���ۏɌW��Ɩ���S������g�D�ɑ��ĕ����ɂ����邱�ƁB
�Q�@�����Ǝғ��́A�O����ꍆ�̃o���f�[�V�����̌��ʂɊ�Â��A�����Ǘ����͕i���Ǘ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i�ύX�̊Ǘ��j
��\�l���@�����Ǝғ��́A�����A���ގႵ���͐��i�̋K�i���͐����菇���ɂ��ĕύX���s���ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�ύX�ɂ�鐻�i�i���y�я��F�����ւ̉e����]�����邱�ƁB
��@�O���̕]���̌��ʂ���A���Y�ύX�����i�i���Ⴕ���͏��F�����ɉe�����y�ڂ��ꍇ���͂��̂����ꂪ����ꍇ�ɂ́A���Y�ύX�Ɋ֘A���鐻�i�ɌW�鐻���̔��Ǝҋy�і@��\����̓��ꍀ�̏��F�����҂ɑ��ĘA�����A�m�F���邱�ƁB
�O�@�O�̕]���y�ъm�F�̌��ʂɊ�Â��A���Y�ύX���s�����Ƃɂ��ĕi���ۏɌW��Ɩ���S������g�D�̏��F���邱�ƁB
�l�@�O���̏��F���ĕύX���s���ɍۂ��āA�֘A���镶���̉����A�E���̋���P�����̑����v�̑[�u���Ƃ邱�ƁB
�܁@�O�e���̋Ɩ��̎��{���A�i���ۏɌW��Ɩ���S������g�D�y�ѐ����Ǘ��҂ɑ��ĕ����ɂ����邱�ƁB
�Z�@�O�e���̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�O���̕ύX���s���������Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���i�i���ւ̉e�����Ċm�F���A���Y�ύX�̖ړI���B������Ă��邱�Ƃ��m�F���邽�߂̕]�����s�����ƁB
��@���i�i�����͏��F�����ɉe�����y�ڂ��ύX���s�����ꍇ�ɂ����ẮA���Y���i�ɌW�鐻���̔��Ǝҋy�і@��\����̓��ꍀ�̏��F�����҂ɑ��ĘA�����邱�ƁB
�O�@�O�̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�i��E�̊Ǘ��j
��\���@�����Ǝғ��́A�����菇������̈�E�i�ȉ��P�Ɂu��E�v�Ƃ����B�j���������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@��E�̓��e���L�^����ƂƂ��ɁA��E�������Ƃɂ��e�������A���̌��ʂɂ��ĕi���ۏɌW��Ɩ���S������g�D�ɑ��ĕ����ɂ����A�m�F���邱�ƁB
��@�d��Ȉ�E���������ꍇ�ɂ����ẮA�O���ɒ�߂���̂̂ق��A���Ɍf����Ɩ����s���ƂƂ��ɁA���̓��e�ɂ��ĕi���ۏɌW��Ɩ���S������g�D�ɑ��ĕ����ɂ����A�m�F���邱�ƁB
�C�@���Y��E�Ɋ֘A���鐻�i�ɌW�鐻���̔��Ǝ҂ɑ��đ��₩�ɘA�����邱�ƁB
���@���Y��E�̌������������邱�ƁB
�n�@���v�̐����[�u�y�ї\�h�[�u���Ƃ邱�ƁB
�O�@�O�̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A�O����ꍆ�y�ё�ɂ��m�F�����L�^���쐬�����A�ۊǂ�����ƂƂ��ɁA�����Ǘ��҂ɑ��ĕ����ɂ��K�ɕ����Ȃ���Ȃ�Ȃ��B
�i�i�����y�ѕi���s�Ǔ��̏����j
��\�Z���@�����Ǝғ��́A���i�ɌW��i�����Ɋւ�����i�ȉ��u�i�����v�Ƃ����B�j���Ƃ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�i�����̓��e���L�ڂ����L�^���쐬���A�����ۊǂ��邱�ƁB
��@���Y�i�����ɌW�鎖�������̐������ɋN��������̂łȂ����Ƃ����炩�ȏꍇ�������A���̌������������A�����E�i���֘A�Ɩ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̐����[�u�y�ї\�h�[�u���Ƃ邱�ƁB
�O�@�O���̌��������̌��ʕ��тɐ����[�u�y�ї\�h�[�u�̋L�^���쐬���A�����ۊǂ���ƂƂ��ɁA�i���ۏɌW��Ɩ���S������g�D�ɑ��ĕ����ɂ�葬�₩�ɕ��A�m�F���邱�ƁB
�l�@�O���̕y�ъm�F�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�O����O���̊m�F�ɂ��i���s�ǖ��͂��̂����ꂪ���������ꍇ�ɂ́A�i���ۏɌW��Ɩ���S������g�D�ɁA�菇�����Ɋ�Â��A���Y�������Ǘ��҂ɑ��ĕ����ɂ������Ȃ���Ȃ�Ȃ��B�܂��A���Y�i�����Ɋ֘A���鐻�i�ɌW�鐻���̔��Ǝ҂ɑ��鑬�₩�ȘA���A���i�̉���̔��f�ɕK�v�ȏ��̒��A���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�ɌW��L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i������̏����j
��\�����@�����Ǝғ��́A������ꂽ���i��ۊǂ���ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@������ꂽ���i���敪���Ĉ����ԕۊǂ�����A�K�ɏ������邱�ƁB
��@������ꂽ���i�̓��e���L�ڂ����ۊNjy�я����̋L�^���쐬���A�����ۊǂ���ƂƂ��ɁA�i���ۏɌW��Ɩ���S������g�D�y�ѐ����Ǘ��҂ɑ��ĕ����ɂ����邱�ƁB
�Q�@�g�p���͏o�ׂɕs�K�Ƃ��ꂽ�����A���ދy�ѐ��i�̕ۊNjy�я����ɂ��ẮA�O���̋K������p����B
�i���ȓ_���j
��\�����@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�����E�i���֘A�Ɩ��ɂ��Ē���I�Ɏ��ȓ_�����s�����ƁB
��@���ȓ_���̌��ʂ�i���ۏɌW��Ɩ���S������g�D�y�ѐ����Ǘ��҂ɑ��ĕ����ɂ����邱�ƁB
�O�@���ȓ_���̌��ʂ̋L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�O����ꍆ�̎��ȓ_���̌��ʂɊ�Â��A�����E�i���֘A�Ɩ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�i����P���j
��\����@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�����E�i���֘A�Ɩ��ɏ]������E���ɑ��āA�����Ǘ��y�ѕi���Ǘ��Ɋւ���K�v�ȋ���P�����v��I�Ɏ��{���邱�ƁB
��@����P���̎��{��i���ۏɌW��Ɩ���S������g�D�y�ѐ����Ǘ��҂ɑ��ĕ����ɂ����邱�ƁB
�O�@����P���̎��{�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�l�@����P���̎����������I�ɕ]�����A�K�v�ɉ����ĉ��P��}��ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�i�����y�ыL�^�̊Ǘ��j
���\���@�����Ǝғ��́A���̏͂ɋK�肷�镶���y�ыL�^�ɂ��āA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�������쐬���A���͉�������ꍇ�ɂ����ẮA���F�A�z�t�A�ۊǓ����s�����ƁB
��@�菇�������쐬���A���͉�������Ƃ��́A���Y�菇�����ɂ��̓��t���L�ڂ���ƂƂ��ɁA����ȑO�̉����ɌW�闚����ۊǂ��邱�ƁB
�O�@���̏͂ɋK�肷�镶���y�ыL�^���A�쐬�̓��i�菇�����ɂ��Ă͎g�p���Ȃ��Ȃ������j����ܔN�ԁi�������A���Y�L�^���ɌW�鐻�i�̗L�����ԂɈ�N�����Z�������Ԃ��ܔN��蒷���ꍇ�ɂ����ẮA����P���ɌW��L�^�������A���̗L�����ԂɈ�N�����Z�������ԁj�ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�菇�����y�т��̏͂ɋK�肷��L�^�ɂ��āA���炩���ߎw�肵���҂ɁA�攪���ɋK�肷�镶���Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�쐬�y�ѕۊǂ��ׂ��菇�������тɋL�^�Ɍ������Ȃ��悤�A�p���I�ɊǗ����邱�ƁB
��@�쐬���ꂽ�菇�����y�ыL�^�����m�ȓ��e�ł���悤�A�p���I�ɊǗ����邱�ƁB
�O�@���̎菇�����y�ыL�^�̓��e�Ƃ̕s�������Ȃ��悤�A�p���I�ɊǗ����邱�ƁB
�l�@�菇�����Ⴕ���͋L�^�Ɍ������������ꍇ���͂��̓��e�ɕs���m�Ⴕ���͕s�����ȓ_�����������ꍇ�ɂ����ẮA���̌������������A���v�̐����[�u�y�ї\�h�[�u���Ƃ邱�ƁB
�܁@���̑��菇�����y�ыL�^�̐M�������m�ۂ��邽�߂ɕK�v�ȋƖ�
�Z�@�O�e���̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
���߁@������i�̐����Ǘ��y�ѕi���Ǘ�
�i�i���Ǘ��j
���\����@������i�̐����Ǝғ��́A���Y���i�ɂ��āA�i������ɁA�菇�����Ɋ�Â��A���b�g���Ƃɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��Q�l�i�Ƃ��āA�������ꂽ������A���̊e���Ɍf������ԓK�ȕۊǏ����̉��ŕۊǂ����Ȃ���Ȃ�Ȃ��B
��@�L�����Ԃɑウ�ă��e�X�g�����ݒ肳��Ă�����i�i������ː����i�������B�j�ɂ����ẮA���̃��e�X�g���܂ł̊��Ԗ��͂��̐���������̏o�ׂ���������������O�N�Ԃ̂����ꂩ��������
��@�O���Ɍf������̈ȊO�̈��i�ɂ����ẮA���̗L�����ԂɈ�N�i���Y���i��������ː����i�ł���ꍇ�͘Z�����͕i�����X�N�}�l�W�����g�Ɋ�Â��K�ȓ����j�����Z��������
�i���萫���j�^�����O�j
���\����̓�@������i�̐����Ǝғ��́A���Y���i�ɂ��āA�i������ɁA�菇�����Ɋ�Â��A���Ɍf������萫���j�^�����O�ɌW��Ɩ����v��I���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�i�����X�N����肵�A�]�����s�������ʂɊ�Â��āA���萫���j�^�����O���s�����i��K�ɑI�肵�A�K�v�ʂ̌��̂��̎悷�邱�ƁB
��@���Y���i�̋K�i�̂����ۑ��ɂ��e�����₷�����ڋy�ѓ��Y�K�i�ɓK�����Ȃ��ꍇ�ɓ��Y���i�̗L�������͈��S���ɉe�����y�ڂ��ƍl�����鍀�ڂ��A���������̍��ڂƂ��đI�肷�邱�ƁB
�O�@��ꍆ�̌��̂�ۊǂ��A�O���̍��ڂɂ��āA�K�ȊԊu�Ŏ����������s�����ƁB
�l�@�O���̎��������̌��ʂɊ�Â��A���Y���i�̕i���ւ̉e����]�����邱�ƁB
�܁@�O�e���̋Ɩ��ɌW��L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@������i�̐����Ǝғ��́A�O����l���̕]���̌��ʂ���A���Y���i�̋K�i�ɓK�����Ȃ��ꍇ���͂��̂����ꂪ����ꍇ�ɂ����ẮA���Y���i�ɌW�鐻���̔��Ǝ҂ɑ��鑬�₩�ȘA���A���i�̉���̔��f�ɕK�v�ȏ��̒��A���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�ɌW��L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i�����y�ыL�^�̕ۊǁj
���\����@�����Ǝғ��́A������i�ɌW�鐻�i������ꍇ�ɂ����ẮA���\���ꍀ��O���̋K��ɂ�����炸�A���̏͂ɋK�肷�镶���y�ыL�^�ł����ē��Y���i�ɌW����̂ɂ��ẮA�쐬�̓��i�菇�����ɂ��Ă͎g�p���Ȃ��Ȃ������j���玟�̊e���Ɍf������ԁi�������A����P���ɌW��L�^�ɂ����ẮA�쐬�̓�����ܔN�ԁj�ۊǂ��Ȃ���Ȃ�Ȃ��B
��@���b�g���\��������i�̂����L�����Ԃɑウ�ă��e�X�g�����ݒ肳��Ă�����̂ɌW�镶���y�ыL�^�ɂ����ẮA���Y�����y�ыL�^�ɌW����i�̃��b�g�̃��e�X�g���܂ł̊��Ԗ��͓��Y���b�g�̐���������̏o�ׂ���������������O�N�Ԃ̂����ꂩ��������
��@�O���Ɍf������̈ȊO�̈��i�ɌW�镶���y�ыL�^�ɂ����ẮA���Y���i�̗L�����ԂɈ�N�����Z��������
��O�߁@���ۈ��i�̐����Ǘ��y�ѕi���Ǘ�
�i���ۈ��i�̐������̍\���ݔ��j
���\�O���@�{�s�K�����\���ꍀ��O���̋敪�̐����Ǝҋy�ю{�s�K����O�\���ꍀ��O���̋敪�̊O�������Ǝ҂̐������̍\���ݔ��́A�����ꍀ�ɋK�肷����̂̂ق��A���ɒ�߂�Ƃ���ɓK��������̂łȂ���Ȃ�Ȃ��B
��@��Ə��̂����A��Ǝ����͍�ƊǗ����́A���ۈ��i�ɌW�鐻�i�̎�ށA�܌`�y�ѐ����H���ɉ����A����̒��x���ێ��Ǘ��ł���\���y�ѐݔ���L���邱�ƁB
��@����̗e��̊�����Ɩ��͖ŋۍ�Ƃ��s����Ǝ��͐�p�ł��邱�ƁB�������A����̗e�킪��������邨���ꂪ�Ȃ��ꍇ�ɂ����ẮA���̌���łȂ��B
�O�@��Ǝ��͎��ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@����̗e��̊����y�ѕۊǂ�K�ɍs�����߂ɕK�v�Ȑݔ���L���邱�ƁB
���@���ۈ��i�ɌW�鐻�i�̎�ނɉ����A���̐����ɕK�v�Ȗŋۑ��u������Ă��邱�ƁB
�n�@���ۑ�����s�����́A�t�B���^�[�ɂ�菈�����ꂽ����ȋ�C�������A���A�K�ȍ����Ǘ����s�����߂ɕK�v�ȍ\���ݔ���L���邱�ƁB
�j�@���ˍ܂ɌW�鐻�i������ꍇ�ɂ����ẮA���ې��ۏɉe�����y�ڂ��ډt���̔z�Ǔ��́A��e�ՂŁA���A�ŋۂ��\�Ȑݔ��ł��邱�ƁB
�l�@��܂̒�����ƁA�[�U��ƁA���͐��i�̖ŋۂ̂��߂ɍs��������ƈȍ~�̍�Ɓi�\���y�ѕ��Ƃ������B�j���s����Ǝ����͍�ƊǗ����́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@�ۈ��i�̍�Ə��Ƌ�ʂ���Ă��邱�ƁB
���@������Ƃ��s����Ǝ��y�я[�U��Ɩ��͕Ǎ�Ƃ��s����Ǝ��͐�p�ł��邱�ƁB
�n�@���̍�Ƃ��s���E���̐�p�̍X�ߎ���L���邱�ƁB
�܁@���ۈ��i�ɌW�鐻�i�̐����ɕK�v�ȏ�����������������ݔ��́A�ٕ����͔������ɂ����������̉�����h�~���邽�߂ɕK�v�ȍ\���ł��邱�ƁB
�i�����Ǘ��j
���\�l���@�����Ǝғ��́A���ۈ��i�ɌW�鐻�i������ꍇ�ɂ����ẮA��������ɁA��\���ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@��Ƌ��ɂ��ẮA�������閳�ۈ��i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H���y�ѓ��Y���ōs����Ɠ��e���ɉ����āA����̒��x����Ɗ��̊Ǘ��̒��x��K�ɐݒ肵�A�Ǘ����邱�ƁB
��@�����A���ދy�ѐ��i�ɂ��ẮA�������閳�ۈ��i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H�����ɉ����āA���������̐����K�v�ȊǗ����ڂ�K�ɐݒ肵�A�Ǘ����邱�ƁB
�O�@�����H���ɂ����āA�����A���ދy�ѐ��i�̔��������ɂ�鉘������h�~���邽�߂ɕK�v�ȑ[�u���Ƃ邱�ƁB
�l�@�������閳�ۈ��i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H�����ɉ����āA���i�̖��ې���ۏ��邽�߂ɏd�v�ȍH�����ɂ��ẮA�H���Ǘ��̂��߂ɕK�v�ȊǗ��l��K�ɒ�߁A�Ǘ����邱�ƁB
�܁@�����p���ɂ��ẮA���̗p�r�ɉ����A���v�̔������w�I���ڋy�ѕ������w�I���ڂɌW��Ǘ��l��K�ɒ�߁A�Ǘ����邱�ƁB
�Z�@���ɒ�߂�Ƃ���ɂ��A�E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���ȊO�̎҂̍�Ə��ւ̗�������ł�����萧�����邱�ƁB
���@�����g�D�����̉��H�A�������̔|�{���i���̐����H���ɂ����Č��Ɍ����y�эޗ��Ƃ��Ďg�p����Ă�����̂������B�j�ɌW���Ƃɏ]������E���ɂ�鉘���̖h�~�̂��߂̌��d�Ȏ菇���߁A��������炷��ꍇ�������A���ۈ��i�ɌW�鐻�i�̍�Ƌ��ɗ����肳���Ȃ����ƁB
�n�@���ɍ�Ƃ��s���Ă��鐴���斔�͖��ۋ��ւ̐E���̗�������ł�����萧�����邱�ƁB
���@���ɒ�߂�Ƃ���ɂ��A�����斔�͖��ۋ��ō�Ƃ���E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���������斔�͖��ۋ��֗�����ۂɂ́A���Y���̊Ǘ��̒��x�ɉ����āA�X�ߓ���K�ɍs�킹�邱�ƁB
���@�E���������A���ދy�ѐ��i����������ɂ�艘�����邨����̂��錒�N��ԁi�畆�Ⴕ���͖є��̊����ǎႵ���͕��ׂɂ������Ă���ꍇ�A�������Ă���ꍇ���͉����Ⴕ���͌����s���̔��M���̏Ǐ��悵�Ă���ꍇ���܂ށB�ȉ������B�j�ɂ���ꍇ�ɂ����ẮA�\�����s�킹�邱�ƁB
�i����P���j
���\���@�����Ǝғ��́A���ۈ��i�ɌW�鐻�i������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA��\����ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�������͎��������ɏ]������E���ɑ��āA���ۈ��i�ɌW�鐻�i�̐����̂��߂ɕK�v�ȉq���Ǘ��A�������w���̑��K�v�ȋ���P�������{���邱�ƁB
��@������y�і��ۋ�擙�ł̍�Ƃɏ]������E���ɑ��āA���������ɂ�鉘����h�~���邽�߂ɕK�v�ȑ[�u�Ɋւ��鋳��P�������{���邱�ƁB
��l�߁@�����R�����i���̐����Ǘ��y�ѕi���Ǘ�
�i�����R�����i���ɌW����i���i�W�����j
���\���̓�@�����Ǝғ��́A�����R�����i�A���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���{�s�߁i���a�O�\�Z�N���ߑ�\�ꍆ�j�攪�\����O���C�Ɍf���鐶���w�I���܁A�@��l�\�O���ꍀ�̋K��ɂ������J����b�̎w�肵�����i�A��`�q�g�����Z�p�����p���Đ����������i�A��`�q�g�����Z�p�����p���Đ����������i�������Ƃ��Ďg�p������i�A�l�Ⴕ���͓����̍זE��|�{����Z�p�����p���Đ����������i�A�l�Ⴕ���͓����̍זE��|�{����Z�p�����p���Đ����������i�������Ƃ��Ďg�p������i���͍זE�g�D���i�i�ȉ��u�����R�����i���v�Ƒ��̂���B�j�ɌW�鐻�i������ꍇ�ɂ����ẮA���i���i�W�����ɁA�掵���ɋK�肷�鎖���̂ق��A���Ɍf���鎖�����L�ڂ��A�i������̏��F������̂Ƃ���ƂƂ��ɁA���Y���i���i�W�����Y�������ɓK�ɔ����u���Ȃ���Ȃ�Ȃ��B
��@�����Ƃ��Ďg�p����l�A�����A�A�����͔��������瓾��ꂽ���ɌW�閼�́A�{���y�ѐ�����тɐ����y�т��̊ܗL�ʂ��̑��̋K�i
��@�������͎��������Ɏg�p���铮���i�h�i�[�������܂ށB�j�i�ȉ��u�g�p�����v�Ƃ����B�j�̋K�i�i����Ǘ��̕��@���܂ށB�j
�i�����R�����i���̐������̍\���ݔ��j
���\�Z���@�����R�����i���ɌW�鐻�i�̐����Ǝғ��̐������̍\���ݔ��́A�����ꍀ�y�ё��\�O���̋K��ɒ�߂���̂̂ق��A���ɒ�߂�Ƃ���ɓK��������̂łȂ���Ȃ�Ȃ��B
��@�����w�I���܁i���b�g���\�����Ȃ����t���܂������B�j�ɌW�鐻�i�̐������̍\���ݔ��́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@��Ə��ɂ́A�����疾�m�ɋ�ʂ��ꂽ���ɂ����āA���Ɍf����ݔ���݂��邱�ƁB�������A���i�̎�ށA�������@���ɂ��A���Y���i�̐����ɕK�v���Ȃ��ƔF�߂���ݔ��������B
�i�P�j�@�������̒����ݔ�
�i�Q�j�@�g�p�����Ŕ������ڎ��̂��̂��Ǘ�����ݔ�
�i�R�j�@�g�p��������������ݔ�
�i�S�j�@��������|�n���ɈڐA����ݔ�
�i�T�j�@��������|�{����ݔ�
�i�U�j�@�|�{�����������̍̎�A�s�����A�E�ۓ����s���ݔ�
�i�V�j�@���t�̊�ߗp�t������ݔ�
�i�W�j�@���t�̊�߁A�����y�їe��̕ǂ��s���ݔ�
�i�X�j�@�������͎��������Ɏg�p��������B���ɂ��ď��ł��s���ݔ�
���@�C�i�S�j�y�сi�U�j����i�W�j�܂łɌf����ݔ���L���鎺���тɌ����A���ދy�ѐ��i�̎��������ɕK�v�Ȑݔ��̂������ێ������s���ݔ���L���鎺�́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�i�P�j�@���ێ��ł��邱�ƁB�������A���Y��Ǝ����ɁA���i�̎�ށA�������@���ɂ��x��Ȃ����ۓI������s�����Ƃ��ł���@�\��L����ݔ���݂���ꍇ�ɂ����ẮA���̌���ł͂Ȃ��B
�i�Q�j�@�i�P�j�̖��ێ��ɂ́A��p�̑O���u���A�ʏ퓖�Y�O����ʂ��Ă̂ݍ�Ǝ����ɏo����ł���悤�ȍ\���̂��̂Ƃ��A���A���̑O���̏o���������O�ɒ��ږʂ��Ă��Ȃ����̂ł��邱�ƁB
�n�@�C�Ɍf������̂̂ق��A���Ɍf����ݔ���L���邱�ƁB
�i�P�j�@�g�p�����̎���Ǘ��ɕK�v�Ȑݔ�
�i�Q�j�@�|�n�y�т��̊�ߗp�t������ݔ�
�i�R�j�@�������͎��������Ɏg�p�������B�A�e�퓙�ɂ��Ă��炩���ߐ��y�іŋۂ��s���ݔ�
�i�S�j�@�����̎��̂��̑��̉����̓K�ȏ����y�щ����̏��s���ݔ�
��@���b�g���\�����Ȃ����t���܂ɌW�鐻�i�̐������̍\���ݔ��́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@��Ə��̂����A���t�����̕����y�э����A��t�̒����y�єr�o���тɗe��̕Ǎ�Ƃ��s����Ǝ��́A���t���܈ȊO�̐��i�̍�Ǝ��Ƌ�ʂ���Ă��邱�ƁB
���@��Ǝ��̂����A�C�ɋK�肷���Ƃ��J��������ɂ���čs����Ǝ��́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�i�P�j�@��Ǝ��͐�p�ł��邱�ƁB
�i�Q�j�@��Ǝ��͖��ێ��ł��邱�ƁA���͓��Y��Ǝ����ɓK�ɖ��ۑ�����s�����Ƃ��ł���@�\��L����ݔ���݂��Ă��邱�ƁB
�n�@��Ə��ɂ́A���ێ��ō�Ƃ��s���E���̐�p�̍X�ߐݔ���݂��邱�ƁB
�O�@�l�̌��t���͌������悤�������Ƃ��鐻�i�̐������s�����́A���̋�悩�疾�m�ɋ�ʂ���Ă���A���A���Y�������s�����߂̐�p�̐ݔ��y�ъ���L���Ă��邱�ƁB�������A�E�C���X��s�������͏�������H���ȍ~�̐����H���ɂ����ẮA���̌���łȂ��B
�i�����Ǘ��j
���\�����@�����Ǝғ��́A�����R�����i���ɌW�鐻�i������ꍇ�ɂ����ẮA��������ɁA��\���y�ё��\�l���ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�����H���ɂ����āA���i����s��������ꍇ���͐��i���Ɋ܂܂�����������s�������A�Ⴕ���͏�������ꍇ�ɂ����ẮA���Y�s�������͏������s���Ă��Ȃ����i���ɂ�鉘����h�~���邽�߂ɕK�v�ȑ[�u���Ƃ邱�ƁB
��@�����H���ɂ����āA���y���̐������w�I�ȋZ�p��p����ꍇ�ɂ����ẮA���x�A���f�C�I���w�����̐����H���̊Ǘ��ɕK�v�Ȏ����ɂ��āA�p���I�ɑ�����s�����ƁB
�O�@�����H���ɂ����āA�J�����N���}�g�O���t���u����p����ꍇ�ɂ����ẮA���������ɂ�铖�Y���u�̉�����h�~���邽�߂ɕK�v�ȑ[�u���Ƃ�ƂƂ��ɁA�K�v�ɉ����G���h�g�L�V���̑�����s�����ƁB
�l�@�����H���ɂ����āA�|�{�����ɘA���I�ɔ|�n���������A���A�A���I�ɔ|�{�t��r�o������|�{������p����ꍇ�ɂ����ẮA�|�{���Ԓ��̓��Y�|�{���ɂ�����|�{�������ێ����邽�߂ɕK�v�ȑ[�u���Ƃ邱�ƁB
�܁@���ɒ�߂�Ƃ���ɂ��A�E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���ȊO�̎҂̍�Ə��ւ̗�������ł�����萧�����邱�ƁB
���@���ɍ�Ƃ��s���Ă��鐴���斔�͖��ۋ��ւ̐E���̗�������ł�����萧�����邱�ƁB
�n�@������Ƃɏ]������E�����A�g�p�����i���̐����H���ɂ����Č��Ɏg�p����Ă�����̂������B�j�̊Ǘ��ɌW���Ƃɏ]�������Ȃ����ƁB
�Z�@���ɒ�߂�Ƃ���ɂ��A�����斔�͖��ۋ��ō�Ƃ���E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���ɁA���ł��ꂽ��ƈ߁A��Ɨp�̂͂����A��ƖX�y�э�ƃ}�X�N�𒅗p�����邱�ƁB
���@�E���������A���ދy�ѐ��i����������ɂ�艘�����邨����̂��鎾�a�ɂ������Ă��Ȃ����Ƃ��m�F���邽�߂ɁA�E���ɑ��A�Z�����Ȃ����Ԃ��ƂɌ��N�f�f���s�����ƁB
�n�@�E���������A���ދy�ѐ��i����������ɂ�艘�����邨����̂��錒�N��Ԃɂ���ꍇ�ɂ����ẮA�\�����s�킹�邱�ƁB
���@�g�p�����i�����Ɏg�p������̂Ɍ���B�ȉ����̍��ɂ����ē����B�j���펞�K���ȊǗ��̉��Ɏ��炷��ƂƂ��ɁA���̎g�p�ɓ������ẮA���N�ώ@���s�����Ƃɂ��A�`���a�ɂ������Ă��铮�����̑��g�p�ɓK���Ă��Ȃ��������g�p���邱�Ƃ̂Ȃ��悤�ɂ��邱�ƁB
���@�������ɂ�艘�����ꂽ���ׂĂ̕��i�i�����̉ߒ��ɂ����ĉ������ꂽ���̂Ɍ���B�j�y�юg�p�����̎��̂��A�ی��q����̎x�Ⴊ�����邨����̂Ȃ��悤�ɏ��u���邱�ƁB
��@�����Ɏg�p����������̊��̎戵���ɂ��āA���Ɍf���鎖���Ɋւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�C�@�������̖��̋y�їe�킲�Ƃɕt���ꂽ�ԍ�
���@���̔N�������тɑ�����̎����y�яZ���i�@�l�ɂ����ẮA���̋y�я��ݒn�j
�n�@�����w�I����y�т��̌����N����
�j�@�p��|�{�̏�
�\�@�������a���́A�}���D�������a���́A�L��E�a���ۖ��͌��j�ۂ���舵����Ǝ��Ŏg�p�������B�́A���i�̎�ނ��ƂɕW����t���āA���̐��i�̐����Ɏg�p���邱�Ƃ��֎~���邱�ƁB
�\��@�����R�����i�ɌW�鐻�i�̐����Ɏg�p���鐶���R�������ɂ��ẮA���Y�����R�����������Y���i�̈��i���i�W�����ɏƂ炵�ēK�Ȃ��̂ł��邱�Ƃ��m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�\��@�����R�����i�ɌW�鐻�i�̐����Ɏg�p���鐶���R�������ɂ��ẮA�����J����b�̒�߂�Ƃ���ɂ��A�L�^���Ȃ���Ȃ�Ȃ��Ƃ���Ă��鎖���̋L�^���A��O�\���ꍆ�y�ё�ɋK�肷����Ԏ���ۊǂ��A���͑�\����̎l��̎挈�߂�������邱�Ƃɂ��A���Y�����R�������̌��ޗ��i�����Ɏg�p���錴�����͍ޗ��i�����H���ɂ����Ďg�p�������̂��܂ށB�j�̗R���ƂȂ���̂������B�j���̎悷��Ǝғ��i�ȉ��u���ޗ��̎�Ǝғ��v�Ƃ����B�j�ɂ����ēK�ɕۊǂ��邱�ƂƂ��邱�ƁB
�\�O�@��\���\���y�ёO�̋L�^���A�������鐶���R�����i�����鐻�i�̃��b�g���Ƃɍ쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�זE�g�D���i�ɌW�鐻�i������ꍇ�ɂ����ẮA��������ɁA��\���y�ёO���ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�قȂ�h�i�[���̓h�i�[��������̎悵���זE���͑g�D����舵���ꍇ�ɂ����ẮA���Y�זE���͑g�D�̍����y�ь���������h�~���邽�߂ɕK�v�ȑ[�u���Ƃ邱�ƁB
��@�����ƂȂ�זE���͑g�D�ɂ��āA����ꎞ�ɁA���Ɍf���鎖���Ɋւ���L�^�ɂ��A���Y���i�̈��i���i�W�����ɏƂ炵�ēK�Ȃ��̂ł��邱�Ƃ��m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�C�@���Y�זE���͑g�D���̎悵���{��
���@���Y�זE���͑g�D���̎悵���N����
�n�@���Y�זE���͑g�D���l�ɌW����̂ł���ꍇ�ɂ����ẮA�h�i�[�X�N���[�j���O�̂��߂̃h�i�[�̖�f�A�������ɂ��f�f�̏�
�j�@���Y�זE���͑g�D�������ɌW����̂ł���ꍇ�ɂ����ẮA�h�i�[�����̎����̏��тɃh�i�[�����X�N���[�j���O�̂��߂̃h�i�[�����̎��������y�ю���Ǘ��̏�
�z�@���Y�זE���͑g�D���̎悷���Ƃ̌o��
�w�@�C����z�܂łɌf������̂̂ق��A�זE�g�D���i�ɌW�鐻�i�̕i���̊m�ۂɊւ��K�v�Ȏ���
�O�@�����ƂȂ�זE���͑g�D���h�i�[��������̎悷��ꍇ�ɂ����ẮA�̎�̉ߒ��ɂ�������������ɂ�鉘����h�~���邽�߂ɕK�v�ȑ[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�l�@�E�������̂����ꂩ�ɊY������ꍇ�ɂ����ẮA���Y�E���𐴏��斔�͖��ۋ��ɂ������Ƃɏ]�������Ȃ����ƁB
�C�@�����A���ދy�ѐ��i����������ɂ�艘�����邨����̂��錒�N��Ԃɂ���ꍇ
���@�זE���͑g�D�̍̎斔�͉��H�̒��O�ɍזE���͑g�D���������邨����̂��������������舵���Ă���ꍇ
�܁@���i�ɂ��āA���i���ƂɁA�o�א�{�ݖ��A�o�ד��y�у��b�g��c������ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�Z�@�z���ɂ��āA���i�i���̊m�ۂ̂��߂ɕK�v�ȑ[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��邱�ƁB
���@�h�i�[�����̎�����̎���Ǘ��Ɋւ���L�^���쐬���A�����ۊǂ��邱�ƁB
���@��A��O���A��܍��y�ё�Z���̋L�^���A���b�g�i��܍��̋L�^�ɂ����ẮA���i�j���Ƃɍ쐬���A�����ۊǂ��邱�ƁB
�R�@��\���y�ёO�ɋK�肷�鐶���R�����i�ɌW�鐻�i�ɌW��L�^�́A�����Ɏg�p���������R�������Ɋւ���L�^���瓖�Y�����R���������g�p���Đ������ꂽ���i�Ɋւ���L�^�܂ł̈�A�̂��̂�K�Ɋm�F�ł���悤�ɕۊǂ���Ȃ���Ȃ�Ȃ��B
�i�i���Ǘ��j
���\�����@�@�����\�ꍀ�ɋK�肷����萶���R�����i������i�i�ȉ��u���萶���R�����i�v�Ƃ����B�j���͍זE�g�D���i�̍ŏI���i�̐����Ǝғ��́A���Y�ŏI���i�ɂ��āA��\����ꍀ��܍��y�ё�Z���̋K��ɂ�����炸�A���b�g���Ɓi���b�g���\�����Ȃ����萶���R�����i�ɂ����ẮA���̐����Ɏg�p���������R�������ɂ��āA���Y�ŏI���i�̐����ԍ����͓��Y�����R�������̃��b�g���Ɓj�ɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��Q�l�i�Ƃ��āA�������ꂽ�����玟�̊e���Ɍf������ԓK�ȕۊǏ����̉��ŕۊǂ��Ȃ���Ȃ�Ȃ��B�������A���b�g���\�����Ȃ����萶���R�����i�̐����Ɏg�p���������R�������ł����Č��ޗ��̎�Ǝғ��Ƃ̊Ԃœ��Y���ޗ��̎�Ǝғ����Q�l�i�����̊e���Ɍf������ԕۊǂ��邱�Ƃ��\����̎l��̋K��ɂ���茈�߂Ă�����̂ɂ��Ă͂��̌���łȂ��A�܂��A���b�g���\��������萶���R�����i���͍זE�g�D���i�̍ŏI���i�ɂ����ẮA���̗L�����ԂɈ�N�i���ː����i�̍ŏI���i�ɂ����ẮA�Z�����͕i�����X�N�}�l�W�����g�Ɋ�Â��K�ȓ����j�����Z�������Ԃ��o�߂�����́A���̐����Ɏg�p���������R�������̕ۊǂ������čŏI���i�̕ۊǂɑウ�邱�Ƃ��ł���B
��@���b�g���\��������萶���R�����i�̍ŏI���i�y�у��b�g���\�����Ȃ����萶���R�����i�̐����Ɏg�p���������R�������ɂ����ẮA���̗L�����Ԃɏ\�N�����Z��������
��@�זE�g�D���i�̍ŏI���i�i�O���Ɍf������̂������B�j�ɂ����ẮA�K�Ȋ���
�Q�@�����Ǝғ��́A�����R�����i���ɌW�鐻�i������ꍇ�ɂ����ẮA�i������ɁA��\����ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf���鎎�������ɌW��Ɩ����v��I���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@���̂̍����y�ь�����������h�~���邽�߂ɁA���̂�K�Ȏ��ʕ\���ɂ��敪���邱�ƁB
��@�i���Ǘ���d�v�ł���A���A���i�ł͎��{���邱�Ƃ��ł��Ȃ����������ɂ��ẮA�����H���̓K�Ȓi�K�Ŏ��{���邱�ƁB
�O�@�g�p�����i���������Ɏg�p������̂Ɍ���B�ȉ����̍��ɂ����ē����B�j���펞�K���ȊǗ��̉��Ɏ��炷��ƂƂ��ɁA���̎g�p�ɓ������ẮA���N�ώ@���s�����Ƃɂ��A�`���a�ɂ������Ă��铮�����̑��g�p�ɓK���Ă��Ȃ��������g�p���邱�Ƃ̂Ȃ��悤�ɂ��邱�ƁB
�l�@�������ɂ�艘�����ꂽ���ׂĂ̕��i�i���������̉ߒ��ɂ����ĉ������ꂽ���̂Ɍ���B�j�y�юg�p�����̎��̂��A�ی��q����̎x�Ⴊ�����邨����̂Ȃ��悤�ɏ��u���邱�ƁB
�܁@���������Ɏg�p����������̊��̎戵���ɂ��āA���Ɍf���鎖���Ɋւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�C�@�������̖��̋y�їe�킲�Ƃɕt���ꂽ�ԍ�
���@���̔N�������тɑ�����̎����y�яZ���i�@�l�ɂ����ẮA���̋y�я��ݒn�j
�n�@�����w�I����y�т��̌����N����
�j�@�p��|�{�̏�
�Z�@�����������ʂ̋L�^���A�������鐶���R�����i���ɌW�鐻�i�̃��b�g���Ƃɍ쐬���A�����ۊǂ��邱�ƁB
�R�@�����Ǝғ��́A�זE�g�D���i�ɌW�鐻�i������ꍇ�ɂ����ẮA�i������ɁA��\����y�ёO���ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf����i���ۏ؋y�ю��������ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�h�i�[�����̎���ꎞ�y�ю�����̎����������s�����Ƃ��̑��K�v�ȋƖ�������s���A���͓��Y�Ɩ��̓��e�ɉ����Ă��炩���ߎw�肵���҂ɍs�킹�邱�ƁB
��@�O���ɋK�肷��Ɩ��̋L�^���쐬���A�����ۊǂ��邱�ƁB
�S�@�O�O���ɋK�肷�鐶���R�����i�ɌW��L�^�́A�����Ɏg�p���������R�������Ɋւ���L�^���瓖�Y�����R���������g�p���Đ������ꂽ���i�Ɋւ���L�^�܂ł̈�A�̂��̂�K�Ɋm�F�ł���悤�ɕۊǂ���Ȃ���Ȃ�Ȃ��B
�i����P���j
���\����@�����Ǝғ��́A�����R�����i���ɌW�鐻�i������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA��\����y�ё��\���ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�����R�����i���̐������͎��������ɏ]������E���ɑ��āA�������w�A��w�y�яb��w���Ɋւ��鋳��P�������{���邱�ƁB
��@���ۋ��y�ѕa������������������舵����擙�ł̍�Ƃɏ]������E���ɑ��āA���������ɂ�鉘����h�~���邽�߂ɕK�v�ȑ[�u�Ɋւ��鋳��P�������{���邱�ƁB
�i�����y�ыL�^�̕ۊǁj
��O�\���@�����Ǝғ��́A�����R�����i���ɌW�鐻�i������ꍇ�ɂ����ẮA���\���ꍀ��O���y�ё��\����̋K��ɂ�����炸�A���̏͂ɋK�肷�镶���y�ыL�^�ł����ē��Y���i�ɌW����̂ɂ��āA�쐬�̓��i�菇�����ɂ��Ă͎g�p���Ȃ��Ȃ������j���玟�̊e���Ɍf������ԁi�������A����P���ɌW��L�^�ɂ����Ă͌ܔN�ԁj�ۊǂ��Ȃ���Ȃ�Ȃ��B
��@���萶���R�����i���͐l�̌��t�����ޗ��Ƃ��Đ�������鐶���R�����i�ɌW�鐻�i�ɂ����ẮA���̗L�����ԂɎO�\�N�����Z��������
��@�����R�����i���͍זE�g�D���i�ɌW�鐻�i�i�O���Ɍf������̂������B�j�ɂ����ẮA���̗L�����Ԃɏ\�N�����Z��������
�O�@�O�Ɍf������̈ȊO�̐��i�ɂ����ẮA�ܔN�ԁi�������A���Y���i�̗L�����ԂɈ�N�����Z�������Ԃ��ܔN��蒷���ꍇ�ɂ����ẮA���̗L�����ԂɈ�N�����Z�������ԁj
��ܐ߁@�G��
�i�L�^�̕ۊǂ̓���j
��O�\����@�O���̋K��ɂ�����炸�A�����Ǝғ��́A�����J����b���w�肷�鐶���R�����i�ɌW�鐻�i������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�O���ɋK�肷��L�^���A�����J����b���w�肷����ԁA�ۊǂ����Ȃ���Ȃ�Ȃ��B�������A���Y�����R�����i�̐����Ɏg�p���鐶���R�������ɌW��L�^�ł����āA��\����̎l��̎挈�߂�������邱�Ƃɂ��A���Y�����R�������̌��ޗ��̎�Ǝғ��ɂ����ē��Y���ԓK�ɕۊǂ��邱�ƂƂ���ꍇ�ɂ����ẮA���̌���łȂ��B
��O�́@��O�i�����Ǝғ��̐������ɂ����鐻���Ǘ��y�ѕi���Ǘ�
���߁@�ʑ�
�i��������y�ѕi������j
��O�\����@�����Ǝғ��́A���������ƂɁA�@��\�����\���ɋK�肷��ӔC�Z�p�Җ��͖@��\�O���̎O��ꍀ�̔F������������̐ӔC�ҎႵ���͓��Y�O�������Ǝ҂����炩���ߎw�肵���ҁi�ȉ��u�ӔC�Z�p�ҁv�Ƒ��̂���B�j�̊ē̉��ɁA��������y�ѕi�������u���Ȃ���Ȃ�Ȃ��B
�Q�@�i������́A�������傩��Ɨ����Ă��Ȃ���Ȃ�Ȃ��B
�i�ӔC�Z�p�ҁj
��O�\�O���@�ӔC�Z�p�҂́A���Ɍf����Ɩ����s��Ȃ���Ȃ�Ȃ��B
��@�����Ǘ��y�ѕi���Ǘ��ɌW��Ɩ��i�ȉ��u�����E�i���Ǘ��Ɩ��v�Ƃ����B�j�����A���̓K�����~���Ȏ��{���}����悤�Ǘ��ē��邱�ƁB
��@�i���s�ǂ��̑����i�i���ɏd��ȉe�����y�Ԃ����ꂪ����ꍇ�ɂ����ẮA���v�̑[�u�����₩�ɂƂ��Ă��邱�Ƌy�т��̐i�����悭���m�F���A�K�v�ɉ����A���P�����v�̑[�u���Ƃ�悤�w�����邱�ƁB
�Q�@�����Ǝғ��́A�ӔC�Z�p�҂��Ɩ����s���ɓ������Ďx����邱�Ƃ��Ȃ��悤�ɂ��Ȃ���Ȃ�Ȃ��B
�i�E���j
��O�\�l���@�����Ǝғ��́A�����E�i���Ǘ��Ɩ���K�����~���Ɏ��{������\�͂�L����ӔC�ҁi�ȉ����̏͂ɂ����ĒP�Ɂu�ӔC�ҁv�Ƃ����B�j���A�������̑g�D�A�K�́A�Ɩ��̎�ޓ��ɉ����A�K�ɒu���Ȃ���Ȃ�Ȃ��B
�Q�@�����Ǝғ��́A�������̑g�D�A�K�͋y�ыƖ��̎�ޓ��ɉ����A�K�Ȑl���̐ӔC�҂�z�u���Ȃ���Ȃ�Ȃ��B
�R�@�����Ǝғ��́A�����E�i���Ǘ��Ɩ���K�Ɏ��{������\�͂�L����l�����\���Ɋm�ۂ��Ȃ���Ȃ�Ȃ��B
�S�@�����Ǝғ��́A�����E�i���Ǘ��Ɩ��ɏ]������E���i�ӔC�Z�p�ҋy�ѐӔC�҂��܂ށB�j�̐Ӗ��y�ъǗ��̐����ɂ��K�ɒ�߂Ȃ���Ȃ�Ȃ��B
�i��O�i���i�W�����j
��O�\���@�����Ǝғ��́A��O�i�ɌW�鐻�i�i���Ԑ��i�������B�j�Ɋւ��Ď��Ɍf���鎖���ɂ��ċL�ڂ��������i�ȉ��u��O�i���i�W�����v�Ƃ����B�j�Y���i�̐����ɌW�鐻�������Ƃɍ쐬���A�i������̏��F���A���Y�������ɓK�ɔ����u���Ȃ���Ȃ�Ȃ��B
��@���F�����̂����A���Y�������ɂ����鐻�����@�A�K�i�y�ю������@���̑��K�v�Ȏ���
��@�@��l�\����̋K��ɂ���߂�ꂽ����̑��Ɋւ���@�ߖ��͂���Ɋ�Â����ߎႵ���͏����̂����i���Ɋւ��鎖��
�O�@�����菇�i��ꍆ�̎����������B�j
�l�@���̑����v�̎���
�i�菇���j
��O�\�Z���@�����Ǝғ��́A���������ƂɁA���Ɍf����菇�ɂ��ċL�ڂ����菇�����쐬���A����Y�������ɓK�ɔ����u���Ȃ���Ȃ�Ȃ��B
��@�\���ݔ��y�ѐE���̉q���Ǘ��Ɋւ���菇
��@�����H���A�����ݔ��A�����A���ދy�ѐ��i�̊Ǘ��Ɋւ���菇
�O�@���������ݔ��y�ь��̂̊Ǘ����̑��K�Ȏ��������̎��{�ɕK�v�Ȏ菇
�l�@����������̏o�ׂ̊Ǘ��Ɋւ���菇
�܁@�o���f�[�V�����Ɋւ���菇
�Z�@��l�\����̕ύX�̊Ǘ��Ɋւ���菇
���@��l�\�O���̈�E�̊Ǘ��Ɋւ���菇
���@��l�\�l���̕i�����y�ѕi���s�Ǔ��̏����Ɋւ���菇
��@��������Ɋւ���菇
�\�@���ȓ_���Ɋւ���菇
�\��@����P���Ɋւ���菇
�\��@�����y�ыL�^�̍쐬�A�����y�ѕۊǂɊւ���菇
�\�O�@���̑��K�����~���Ȑ����E�i���Ǘ��Ɩ��ɕK�v�Ȏ菇
�i�\���ݔ��j
��O�\�����@��O�i�ɌW�鐻�i�̐������̍\���ݔ��́A���ɒ�߂�Ƃ���ɓK��������̂łȂ���Ȃ�Ȃ��B
��@��O�i���i�W�����y�ю菇���i�ȉ����̏͂ɂ����āu�菇�����v�Ƒ��̂���B�j�Ɋ�Â��A���̗p�r�ɉ����K�ɐ��|�y�ѕێ炪�s���A�K�v�ɉ����ŋۂ���A�܂��A���̋L�^���쐬����A�ۊǂ���Ă��邱�ƁB
��@���i���ɂ��L�ŃK�X����舵���ꍇ�ɂ����ẮA���̏����ɗv����ݔ���L���邱�ƁB
�O�@��Ə��̂�����Ǝ��́A���i�̎�ށA�܌`�y�ѐ����H���ɉ����A�������͔������ɂ�鉘����h�~����̂ɕK�v�ȍ\���y�ѐݔ���L���Ă��邱�ƁB�������A�����ݔ����̗L����@�\�ɂ�肱��Ɠ����x�̌��ʂ���ꍇ�ɂ����ẮA���̌���łȂ��B
�l�@��Ə��̂����A�����̔��ʍ�ƁA���i�̒�����ƁA�[�U��Ɩ��͕Ǎ�Ƃ��s����Ǝ��́A���Y��Ǝ��̐E���ȊO�̎҂̒ʘH�ƂȂ�Ȃ��悤�ɑ����Ă��邱�ƁB�������A���Y��Ǝ��̐E���ȊO�̎҂ɂ�鐻�i�ւ̉����̂����ꂪ�Ȃ��ꍇ�ɂ����ẮA���̌���łȂ��B
�܁@���i�̐����ɕK�v�Ȏ��y�їʂ̐��i�ݔ��y�ъ����тɗe��̐���܂ށB�j����������ݔ���L���邱�ƁB
�i�����Ǘ��j
��O�\�����@�����Ǝғ��́A��������ɁA�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@�����w�}�����쐬���A�����ۊǂ��邱�ƁB
��@�����w�}���Ɋ�Â��A���i�̐�����Ƃ��s�����ƁB
�O�@�����Ɋւ���L�^�����b�g���Ƃɍ쐬���A�����ۊǂ��邱�ƁB
�l�@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɁA���ꂪ�K���ł���|���m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
�܁@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɓK���ɕۊǂ��A�o�[���s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�Z�@�\���ݔ��̐�����m�F����ƂƂ��ɁA���̌��ʂɊւ���L�^���쐬���A�����ۊǂ��邱�ƁB
���@�E���̉q���Ǘ����s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
���@�\���ݔ������I�ɓ_����������ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB�܂��A�v��̍Z����K�ɍs���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@�����A�ۊNjy�яo�[���тɉq���Ǘ��Ɋւ���L�^�ɂ�萻���Ǘ����K�ɍs���Ă��邱�Ƃ��m�F���A���̌��ʂ�i������ɑ��ĕ����ɂ����邱�ƁB
�\�@���̑������Ǘ��̂��߂ɕK�v�ȋƖ�
�i�i���Ǘ��j
��O�\����@�����Ǝғ��́A�i������ɁA�菇�����Ɋ�Â��A���Ɍf����i���Ǘ��ɌW��Ɩ����v��I���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɎ����������s���̂ɕK�v�Ȍ��̂��̎悷��ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@�̎悵�����̂ɂ��āA���i���ɂ��Ă̓��b�g���ƂɁA���ނɂ��Ă͊Ǘ��P�ʂ��ƂɎ��������i���Y�����Ǝғ��̑��̎��������ݔ����͑��̎��������@�ւ𗘗p���Ď��Ȃ̐ӔC�ɂ����čs�����������ł����āA���Y���p�ɂ��x�Ⴊ�Ȃ��ƔF�߂�����̂��܂ށB�ȉ����̏͂ɂ����ē����B�j���s���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�O�@�ŏI���i�i���b�g���\��������̂Ɍ���B�j�ɂ��āA���b�g���Ƃɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��Q�l�i�Ƃ��āA�������ꂽ�����瓖�Y���i�̗L�����ԂɈ�N�����Z�������ԓK�ȕۊǏ����̉��ŕۊǂ��邱�ƁB
�l�@���������Ɋւ���ݔ��y�ъ������I�ɓ_����������ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB�܂��A���������Ɋւ���v��̍Z����K�ɍs���ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
�܁@��̎��������̌��ʂ̔�����s���A���̌��ʂ�����ɑ��ĕ����ɂ����邱�ƁB
�Z�@���̑��i���Ǘ��̂��߂ɕK�v�ȋƖ�
�Q�@�A���捑�ɂ����鐻���Ǘ��y�ѕi���Ǘ��̊���тɂ����̊�ɑ���K�����̊m�F�Ɋւ���葱���䂪���̂��̂Ɠ����ł���ƔF�߂���ꍇ�ɂ����ẮA�����Ǝ҂́A�A�����i�ɌW��O����ɋK�肷�鎎�������i�O�ό����������B�j���A���Y�A�����i�ɂ��ėA���捑�̊O�������Ǝ҂��s�������������̋L�^���m�F���邱�Ƃ������đウ�邱�Ƃ��ł���B���̏ꍇ�ɂ����āA�����Ǝ҂́A�i������ɁA���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�A�����i���A���Y�O�������Ǝ҂̐������ɂ����āA�K�Ȑ����菇���ɂ�萻������Ă��邱�Ƃ����I�Ɋm�F���邱�ƁB
��@���Y�O�������Ǝ҂̐��������A���̍��ɂ����鐻���Ǘ��y�ѕi���Ǘ��Ɋւ����ɓK�����Ă��邱�Ƃ����I�Ɋm�F���邱�ƁB
�O�@�O�̊m�F�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�l�@���Y�A�����i�ɂ��ē��Y�O�������Ǝ҂��s�������������̋L�^���m�F����ƂƂ��ɁA���̊m�F�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�R�@�����Ǝғ��́A�i������ɁA�菇�����Ɋ�Â��A�O���㍆�̋K��ɂ�萻�����傩����ꂽ�����Ǘ��ɌW��m�F�̌��ʂ����b�g���ƂɊm�F�����Ȃ���Ȃ�Ȃ��B
�i����������̏o�ׂ̊Ǘ��j
��l�\���@�����Ǝғ��́A�i������ɁA�菇�����Ɋ�Â��A�����Ǘ��y�ѕi���Ǘ��̌��ʂ�K�ɕ]�����A���i�̐���������̏o�ׂ̉ۂ����肷��Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
�Q�@�O���̋Ɩ����s���҂́A���Y�Ɩ���K�����~���Ɏ��{������\�͂�L����҂łȂ���Ȃ�Ȃ��B
�R�@�����Ǝғ��́A��ꍀ�̋Ɩ����s���҂����Y�Ɩ����s���ɓ������āA�x�Ⴊ�����邱�Ƃ��Ȃ��悤�ɂ��Ȃ���Ȃ�Ȃ��B
�S�@�����Ǝғ��́A��ꍀ�̌��肪�K���ɍs����܂Ő��������琻�i���o�ׂ��Ă͂Ȃ�Ȃ��B
�i�o���f�[�V�����j
��l�\����@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Ɍf����ꍇ�ɂ����ăo���f�[�V�������s�����ƁB
�C�@���Y�������ɂ����ĐV���Ɉ�O�i�̐������J�n����ꍇ
���@�����菇���ɂ��Đ��i�i���ɑ傫�ȉe�����y�ڂ��ύX������ꍇ
�n�@���̑����i�̐����Ǘ��y�ѕi���Ǘ���K�ɍs�����ߕK�v�ƔF�߂���ꍇ
��@�o���f�[�V�����̌v��y�ь��ʂ�i������ɑ��ĕ����ɂ����邱�ƁB
�Q�@�����Ǝғ��́A�O����ꍆ�̃o���f�[�V�����̌��ʂɊ�Â��A�����Ǘ����͕i���Ǘ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��Ȃ���Ȃ�Ȃ��B
�i�ύX�̊Ǘ��j
��l�\����@�����Ǝғ��́A�����菇���ɂ��ĕύX���s���ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�ύX�ɂ�鐻�i�i���ւ̉e����]�����A���̕]���̌��ʂ���A���Y�ύX�����i�i���ɉe�����y�ڂ��ꍇ���͂��̂����ꂪ����ꍇ�ɂ́A���Y�ύX���s�����Ƃɂ��ĕi������̏��F����ƂƂ��ɁA���̋L�^���쐬���A�����ۊǂ��邱�ƁB
��@�O���̋K��ɂ��i������̏��F���ĕύX���s���Ƃ��́A�֘A���镶���̉����A�E���̋���P�����̑����v�̑[�u���Ƃ邱�ƁB
�i��E�̊Ǘ��j
��l�\�O���@�����Ǝғ��́A��E���������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@��E�̓��e���L�^���邱�ƁB
��@�d��Ȉ�E���������ꍇ�ɂ����ẮA���Ɍf����Ɩ����s�����ƁB
�C�@��E�ɂ�鐻�i�i���ւ̉e����]�����A���v�̑[�u���Ƃ邱�ƁB
���@�C�ɋK�肷��]���̌��ʋy�ё[�u�ɂ��ċL�^���쐬���A�ۊǂ���ƂƂ��ɁA�i������ɑ��ĕ����ɂ����邱�ƁB
�n�@���̋K��ɂ����ꂽ�]���̌��ʋy�ё[�u�ɂ��āA�i������̊m�F���邱�ƁB
�Q�@�����Ǝғ��́A�i������ɁA�菇�����Ɋ�Â��A�O����n�ɂ��m�F�����L�^���쐬�����A�ۊǂ�����ƂƂ��ɁA�������̋L�^�ƂƂ��ɁA�ӔC�Z�p�҂ɑ��ĕ����ɂ��K�ɕ����Ȃ���Ȃ�Ȃ��B
�i�i�����y�ѕi���s�Ǔ��̏����j
��l�\�l���@�����Ǝғ��́A���i�ɌW��i�������Ƃ��́A���̕i�����ɌW�鎖�������Y�������ɋN��������̂łȂ����Ƃ����炩�ȏꍇ�������A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@���Y�i�����ɌW�鎖���̌������������A�����E�i���Ǘ��Ɩ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̑[�u���Ƃ邱�ƁB
��@���Y�i�����̓��e�A���������̌��ʋy�щ��P�[�u���L�ڂ����L�^���쐬���A�����ۊǂ���ƂƂ��ɁA�i������ɑ��ĕ����ɂ�葬�₩�ɕ��邱�ƁB
�O�@�O���̕ɂ��A�i������̊m�F���邱�ƁB
�Q�@�����Ǝғ��́A�O����O���̊m�F�ɂ��i���s�ǖ��͂��̂����ꂪ���������ꍇ�ɂ́A�i������ɁA�菇�����Ɋ�Â��A���Y������ӔC�Z�p�҂ɑ��ĕ����ɂ������Ȃ���Ȃ�Ȃ��B
�i��������j
��l�\���@�����Ǝғ��́A������ꂽ���i��ۊǂ���ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@������ꂽ���i���敪���Ĉ����ԕۊǂ�����A�K�ɏ������邱�ƁB
��@������ꂽ���i�̓��e���L�ڂ����ۊNjy�я����̋L�^���쐬���A�����ۊǂ���ƂƂ��ɁA�i������y�ѐӔC�Z�p�҂ɑ��ĕ����ɂ����邱�ƁB�������A���Y����Ɏ��������R�����Y�������ɋN��������̂łȂ����Ƃ����炩�ȏꍇ�ɂ����ẮA���̌���łȂ��B
�i���ȓ_���j
��l�\�Z���@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�����E�i���Ǘ��Ɩ��ɂ��Ē���I�Ɏ��ȓ_�����s�����ƁB
��@���ȓ_���̌��ʂ�ӔC�Z�p�҂ɑ��ĕ����ɂ����邱�ƁB
�O�@���ȓ_���̌��ʂ̋L�^���쐬���A�����ۊǂ��邱�ƁB
�Q�@�����Ǝғ��́A�O����ꍆ�̎��ȓ_���̌��ʂɊ�Â��A�����E�i���Ǘ��Ɩ��Ɋւ����P���K�v�ȏꍇ�ɂ����ẮA���v�̑[�u���Ƃ�ƂƂ��ɁA���Y�[�u�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�i����P���j
��l�\�����@�����Ǝғ��́A���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�����E�i���Ǘ��Ɩ��ɏ]������E���ɑ��āA�����Ǘ��y�ѕi���Ǘ��Ɋւ���K�v�ȋ���P�����v��I�Ɏ��{���邱�ƁB
��@����P���̎��{��ӔC�Z�p�҂ɑ��ĕ����ɂ����邱�ƁB
�O�@����P���̎��{�̋L�^���쐬���A�����ۊǂ��邱�ƁB
�i�����y�ыL�^�̊Ǘ��j
��l�\�����@�����Ǝғ��́A���̏͂ɋK�肷�镶���y�ыL�^�ɂ��āA���炩���ߎw�肵���҂ɁA�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�������쐬���A���͉�������ꍇ�ɂ����ẮA���F�A�z�t�A�ۊǓ����s�����ƁB
��@�菇�������쐬���A���͉�������Ƃ��́A���Y�菇�����ɂ��̓��t���L�ڂ���ƂƂ��ɁA����ȑO�̉����ɌW�闚����ۊǂ��邱�ƁB
�O�@���̏͂ɋK�肷�镶���y�ыL�^���A�쐬�̓��i�菇�����ɂ��Ă͎g�p���Ȃ��Ȃ������j����ܔN�ԁi�������A���Y�L�^���ɌW�鐻�i�̗L�����ԂɈ�N�����Z�������Ԃ��ܔN��蒷���ꍇ�ɂ����ẮA����P���ɌW��L�^�������A���̗L�����ԂɈ�N�����Z�������ԁj�ۊǂ��邱�ƁB
���߁@��O�i�̐����̗p�ɋ�����錴��̐����Ǘ��y�ѕi���Ǘ�
�i�i���Ǘ��j
��l�\����@��O�i�̐����̗p�ɋ�����錴��̐����Ǝғ��́A���Y���i�ɂ��āA�i������ɁA�菇�����Ɋ�Â��A���b�g���Ƃɏ���̎��������ɕK�v�ȗʂ̓�{�ȏ�̗ʂ��Q�l�i�Ƃ��āA�������ꂽ������A���̊e���Ɍf������ԓK�ȕۊǏ����̉��ŕۊǂ����Ȃ���Ȃ�Ȃ��B
��@�L�����Ԃɑウ�ă��e�X�g�����ݒ肳��Ă��鐻�i�ɂ����ẮA���̐���������̏o�ׂ���������������O�N��
��@�O���Ɍf������̈ȊO�̐��i�ɂ����ẮA���̗L�����ԂɈ�N�����Z��������
�i�����y�ыL�^�̕ۊǁj
��\���@�����Ǝғ��́A��O�i�̐����̗p�ɋ�����錴��ɌW�鐻�i������ꍇ�ɂ����ẮA��l�\�����O���̋K��ɂ�����炸�A���̏͂ɋK�肷�镶���y�ыL�^�ł����ē��Y���i�ɌW����̂ɂ��ẮA�쐬�̓��i�菇�����ɂ��Ă͎g�p���Ȃ��Ȃ������j���玟�̊e���Ɍf������ԁi�������A����P���ɌW��L�^�ɂ����ẮA�쐬�̓�����ܔN�ԁj�ۊǂ��Ȃ���Ȃ�Ȃ��B
��@���b�g���\�����鐻�i�̂����L�����Ԃɑウ�ă��e�X�g�����ݒ肳��Ă�����̂ɌW�镶���y�ыL�^�ɂ����ẮA���Y�����y�ыL�^�ɌW�郍�b�g�̐���������̏o�ׂ���������������O�N��
��@�O���Ɍf������̈ȊO�̐��i�ɌW�镶���y�ыL�^�ɂ����ẮA���Y���i�̗L�����ԂɈ�N�����Z��������
��O�߁@���ۈ�O�i�̐����Ǘ��y�ѕi���Ǘ�
�i���ۈ�O�i�̐������̍\���ݔ��j
��\����@�{�s�K�����\����ꍆ�̋敪�̐����Ǝҋy�ю{�s�K����O�\����ꍆ�̋敪�̊O�������Ǝ҂̐������̍\���ݔ��́A��O�\�����ɋK�肷����̂̂ق��A���ɒ�߂�Ƃ���ɓK��������̂łȂ���Ȃ�Ȃ��B
��@��Ə��̂����A��Ǝ����͍�ƊǗ����́A���ۈ�O�i�ɌW�鐻�i�̎�ށA�܌`�y�ѐ����H���ɉ����A����̒��x���ێ��Ǘ��ł���\���y�ѐݔ���L���邱�ƁB
��@����̗e��̊�����Ɩ��͖ŋۍ�Ƃ��s����Ǝ��͐�p�ł��邱�ƁB�������A����̗e�킪��������邨���ꂪ�Ȃ��ꍇ�ɂ����ẮA���̌���łȂ��B
�O�@��Ǝ��͎��ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@����̗e��̊����y�ѕۊǂ�K�ɍs�����߂ɕK�v�Ȑݔ���L���邱�ƁB
���@���ۈ�O�i�ɌW�鐻�i�̎�ނɉ����A���̐����ɕK�v�Ȗŋۑ��u������Ă��邱�ƁB
�n�@���ۑ�����s�����́A�t�B���^�[�ɂ�菈�����ꂽ����ȋ�C�������A���A�K�ȍ����Ǘ����s�����߂ɕK�v�ȍ\���ݔ���L���邱�ƁB
�l�@��܂̒�����ƁA�[�U��ƁA���͐��i�̖ŋۂ̂��߂ɍs��������ƈȍ~�̍�Ɓi�\���y�ѕ��Ƃ������B�j���s����Ǝ����͍�ƊǗ����́A���ɒ�߂�Ƃ���ɓK��������̂ł��邱�ƁB
�C�@�ۈ�O�i�̍�Ə��Ƌ�ʂ���Ă��邱�ƁB
���@������Ƃ��s����Ǝ��y�я[�U��Ɩ��͕Ǎ�Ƃ��s����Ǝ��͐�p�ł��邱�ƁB
�n�@���̍�Ƃ��s���E���̐�p�̍X�ߎ���L���邱�ƁB
�܁@���ۈ�O�i�ɌW�鐻�i�̐����ɕK�v�ȏ�����������������ݔ��́A�ٕ����͔������ɂ����������̉�����h�~���邽�߂ɕK�v�ȍ\���ł��邱�ƁB
�i�����Ǘ��j
��\����@�����Ǝғ��́A���ۈ�O�i�ɌW�鐻�i������ꍇ�ɂ����ẮA��������ɁA��O�\�����ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf���鐻���Ǘ��ɌW��Ɩ���K�ɍs�킹�Ȃ���Ȃ�Ȃ��B
��@��Ƌ��ɂ��ẮA�������閳�ۈ�O�i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H���y�ѓ��Y���ōs����Ɠ��e���ɉ����āA����̒��x����Ɗ��̊Ǘ��̒��x��K�ɐݒ肵�A�Ǘ����邱�ƁB
��@�����A���ދy�ѐ��i�ɂ��ẮA�������閳�ۈ�O�i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H�����ɉ����āA���������̐����K�v�ȊǗ����ڂ�K�ɐݒ肵�A�Ǘ����邱�ƁB
�O�@�����H���ɂ����āA�����A���ދy�ѐ��i�̔��������ɂ�鉘������h�~���邽�߂ɕK�v�ȑ[�u���Ƃ邱�ƁB
�l�@�������閳�ۈ�O�i�ɌW�鐻�i�̎�ށA�܌`�A�����A�����H�����ɉ����āA���i�̖��ې���ۏ��邽�߂ɏd�v�ȍH�����ɂ��ẮA�H���Ǘ��̂��߂ɕK�v�ȊǗ��l��K�ɒ�߁A�Ǘ����邱�ƁB
�܁@�����p���ɂ��ẮA���̗p�r�ɉ����A���v�̔������w�I���ڋy�ѕ������w�I���ڂɌW��Ǘ��l��K�ɒ�߁A�Ǘ����邱�ƁB
�Z�@���ɒ�߂�Ƃ���ɂ��A�E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���ȊO�̎҂̍�Ə��ւ̗�������ł�����萧�����邱�ƁB
���@���ɍ�Ƃ��s���Ă��鐴���斔�͖��ۋ��ւ̐E���̗�������ł�����萧�����邱�ƁB
���@���ɒ�߂�Ƃ���ɂ��A�����斔�͖��ۋ��ō�Ƃ���E���̉q���Ǘ����s�����ƁB
�C�@������Ƃɏ]������E���������斔�͖��ۋ��֗�����ۂɂ́A���Y���̊Ǘ��̒��x�ɉ����āA�X�ߓ���K�ɍs�킹�邱�ƁB
���@�E���������A���ދy�ѐ��i����������ɂ�艘�����邨����̂��錒�N��Ԃɂ���ꍇ�ɂ����ẮA�\�����s�킹�邱�ƁB
�i����P���j
��\�O���@�����Ǝғ��́A���ۈ�O�i�ɌW�鐻�i������ꍇ�ɂ����ẮA���炩���ߎw�肵���҂ɁA��l�\�����ɋK�肷��Ɩ��̂ق��A�菇�����Ɋ�Â��A���Ɍf����Ɩ����s�킹�Ȃ���Ȃ�Ȃ��B
��@�������͎��������ɏ]������E���ɑ��āA���ۈ�O�i�ɌW�鐻�i�̐����̂��߂ɕK�v�ȉq���Ǘ��A�������w���̑��K�v�ȋ���P�������{���邱�ƁB
��@������y�і��ۋ�擙�ł̍�Ƃɏ]������E���ɑ��āA���������ɂ�鉘����h�~���邽�߂ɕK�v�ȑ[�u�Ɋւ��鋳��P�������{���邱�ƁB
���@��
�i�{�s�����j
�����@���̏ȗ߂́A�����\���N�l���������{�s����B
�i�o�ߑ[�u�j
�����@�O�������Ǝ҂ɂ��ẮA���̏ȗ߂̎{�s�̓������N�Ԃ́A���̏ȗ߂ɂ�������̑����A���\�O���A���\�Z����тɑ�O�\����ɂ����ď��p��������y�ё��\�O���̋K���K�p���Ȃ����Ƃ��ł���B
��O���@���i�y�ш�O�i�̗A���̔��Ǘ��y�ѕi���Ǘ��K���i�����\��N�����ȗߑ�Z�\�j�͕����\���N�O���O�\�������A���̌��͂������B
���@���@�i������Z�N�����O�Z�������J���ȗߑ攪�����j�@��
�i�{�s�����j
�����@���̏ȗ߂́A�@���̈ꕔ����������@���i�ȉ��u�����@�v�Ƃ����B�j�̎{�s�̓��i������\�Z�N�\�ꌎ��\�ܓ��j����{�s����B
���@���@�i�ߘa�O�N�ꌎ���������J���ȗߑ��܍��j�@��
�i�{�s�����j
�����@���̏ȗ߂́A���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@�����̈ꕔ����������@���i�ȉ��u�����@�v�Ƃ����B�j���������ɋK�肷��K��̎{�s�̓��i�ߘa�O�N��������j����{�s����B
���@���@�i�ߘa�O�N�l���������J���ȗߑ��Z���j�@��
�i�{�s�����j
�����@���̏ȗ߂́A�ߘa�O�N�����������{�s����B
GMP�ȗ߉����ɂ���
�@GMP�ȗ߂Ƃ́A���i����ш�O�i�̐����̔����F�̗v���Ƃ��āA���i����ш�O�i�̐������ɂ����鐻���Ǘ�����ѕi���Ǘ��̊���߂������J���ȗ��ł���B
�@���{�ł́A�u���i�̐����y�ѕi���Ǘ��Ɋւ����v�̒ʒm���o�āA1980�N��GMP�ȗ߂����z���ꂽ�B�ȍ~�A�����J���Ȃ́A���i�����Ɋւ���GMP�̎w�j�����I�ɔ��s���Ă���B
�@GMP�͎���ƂƂ��ɉ�������邽�߁A��ɐV����GMP�ȗ߂ɓK������悤�����̎菇���������������Ƃ��K�v�ł���B
�@�ŋ߂ł́A�ŐV�̍��ەW���ł���PIC/S GMP�K�C�h���C���Ƃ̐��������Ƃ邽�߁AGMP�ȗ߂���16�N�Ԃ�ɉ�������2021�N3���Ɍ��z�A2021�N8��1������{�s���ꂽ�B

�@�����̃|�C���g�͇@�i���ۏ̏[���A�A�O���[�o���Ή��A�B�����̕s���������⏳�F���Ƃ̐������m�ۂ̂R�ŁA�u�i�����X�N�}�l�W�����g�v������u���i�i���V�X�e���v����������A��茵�i�Ȋ�ƂȂ����B
�@
�d�q���{�̑��������ie-Gov�j��GMP�ȗ߁i�����j��������