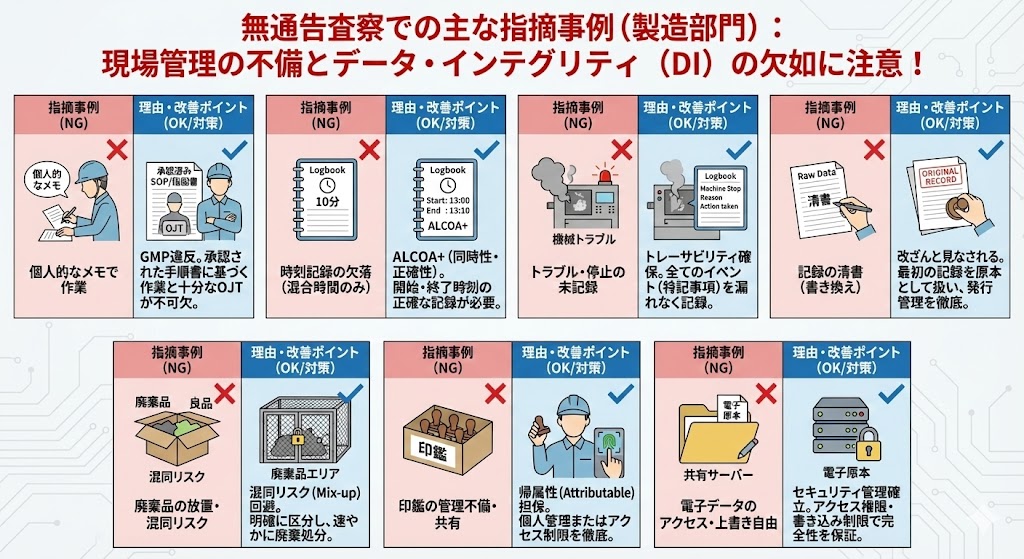
無通告査察での指摘事例(製造部門)
無通告査察において、製造部門では主に現場管理の不備やデータ・インテグリティ(DI)の欠如について、以下のような指摘がなされている。
![]()
【製造領域における主な指摘事例と解説】
| 事例1 |
製造作業担当者が、正式な製造指図記録書ではなく、個人的なメモを参照しながら作業を行っていた。 |
| 理由 |
承認された手順書(SOP)および製造指図書に基づかない作業は、GMP違反である。これは、現行の手順書が実作業に即していない、あるいは作業者へのOJT(教育訓練)が不足していることを示唆しており、手順書の改訂または再教育が不可欠である。 |
| 事例2 |
混合工程において、記録書に「混合時間:10分」とのみ記載されており、実施した「時刻(開始・終了時刻)」の記録が欠落していた。 |
| 理由 |
ALCOA+の原則(同時性・正確性)に基づき、作業の実態を証明するためには、「10分」という結果だけでなく、「13:00〜13:10」といった正確な時刻の記録が必要である。時刻記録がない場合、作業が実際に行われたかの証明が困難となる。 |
| 事例3 |
製造作業中に機械トラブル等による停止が発生したが、その理由、停止期間、および処置内容が製造記録に記載されていなかった。 |
| 理由 |
製造工程における全てのイベント(特記事項)は、トレーサビリティ確保のために漏れなく記録されなければならない。記録の欠落は、逸脱の隠蔽や品質評価の誤りにつながる重大なリスクである。 |
| 事例4 |
現場で採取した「生の記録(Raw Data)」を、照査や査察での体裁を整える目的で、別の様式に清書(書き換え)していた。 |
| 理由 |
最初の記録こそが「原本(Original Record)」であり、これを破棄または書き換える行為はデータの改ざんと見なされる。記録用紙の発行管理を徹底し、現場で記入された記録をそのまま正本として扱う必要がある。 |
| 事例5 |
廃棄すべき品目(廃棄品)が、処分されずに長期間放置されていた。 |
| 理由 |
廃棄品と良品(適合品)の混同リスク(Mix-up)が生じる。不適合品や廃棄対象品は、明確に区分されたエリア(隔離場所)へ移動し、速やかに廃棄処分する手順を順守しなければならない。 |
| 事例6 |
作業者の訂正印や認印が一箇所にまとめて保管されており、他人の印鑑を誰でも自由に使用・持ち出しできる状態であった。 |
| 理由 |
記録の「帰属性(Attributable)」が担保されない。印鑑は個人の署名と同義であり、本人以外が使用できないよう、個人管理を徹底するか、アクセス制限のある保管方法をとる必要がある。 |
| 事例7 |
承認済み製造指図書の電子原本が共有サーバーに保管されていたが、アクセス権限の設定が不十分で、誰でもアクセス・印刷が可能であり、かつ上書き保存もできる状態であった。 |
| 理由 |
未承認の指図書が発行される、あるいは内容が意図せず改変されるリスクがある。電子データのセキュリティ管理(アクセス権限、書き込み制限)を確立し、原本の完全性を保証しなければならない。 |
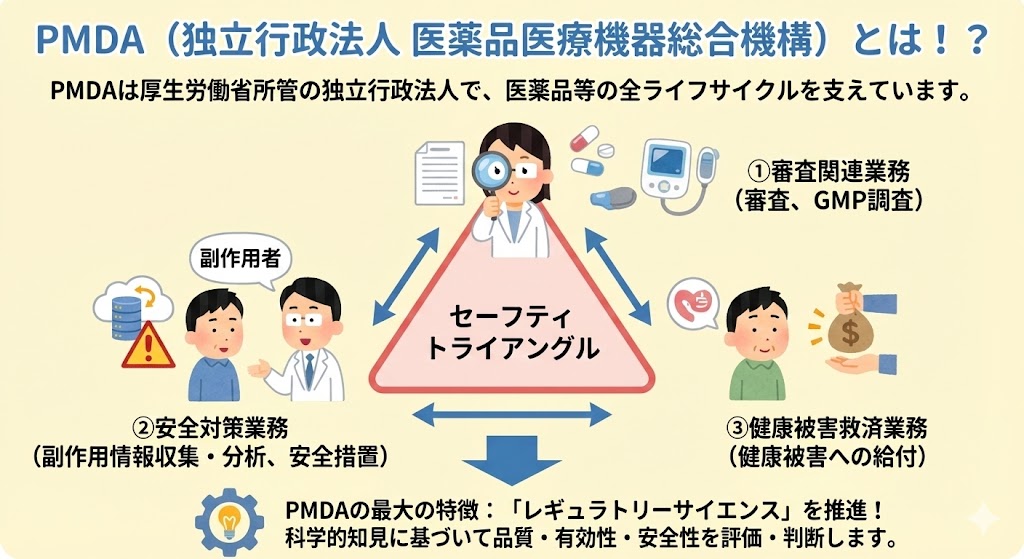
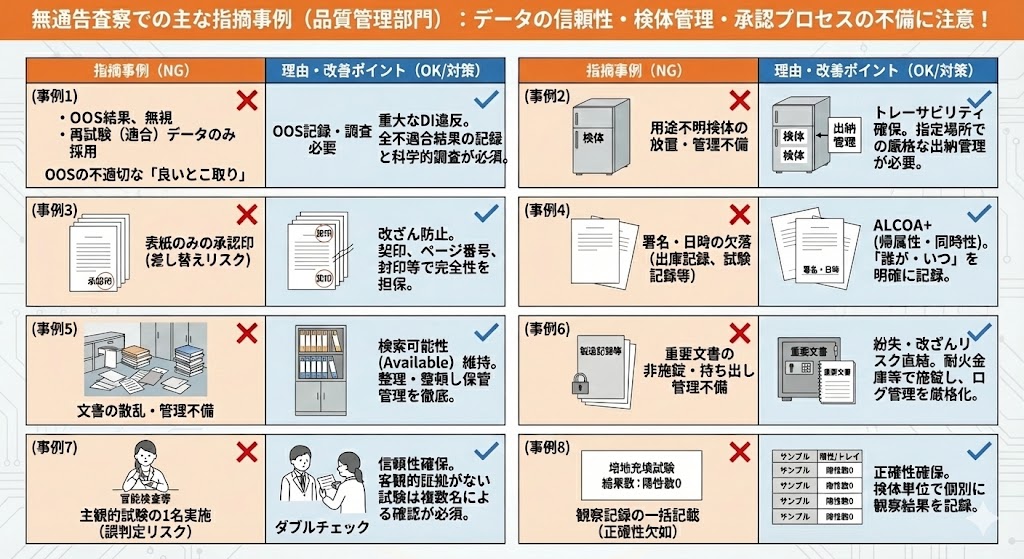
無通告査察での指摘事例(品質管理部門)
品質管理(QC)および品質保証(QA)領域では、データの信頼性、検体管理、承認プロセスに関する指摘が目立つ。
![]()
【品質管理領域における主な指摘事例と解説】
| 事例1 |
初回試験で規格外(OOS)結果が出たにもかかわらず、正規のOOS処理を行わず、試験をやり直して規格内(適合)データのみを採用していた(Testing into Compliance)。 |
| 理由 |
「良いとこ取り」のデータ採用は、重大なDI違反(不正)である。全ての不適合結果はOOSとして記録・調査されなければならず、科学的な正当性なしにデータを破棄・再試験することは認められない。 |
| 事例2 |
用途不明かつ出納管理台帳に記載のない検体が、管理されていない冷蔵庫内に放置されていた。 |
| 理由 |
由来不明の検体が試験に使用される、あるいは意図しない再試験(試し打ち)に悪用されるリスクがある。全ての検体はトレーサビリティを確保し、指定された保管場所で厳格に出納管理を行う必要がある。 |
| 事例3 |
複数ページにわたる指図記録書や試験記録書を発行する際、QA部門が表紙のみに発行印(承認印)を押印していた。 |
| 理由 |
ページの差し替えや抜き取りによる改ざんを防止できない。全ページへの契印(割り印)、ページ番号の付番、あるいは結束して封印するなど、記録の完全性と非複製性を担保するシステムが必要である。 |
| 事例4 |
作業記録において、以下の署名および日時の欠落が多数確認された。
・製品出庫記録における担当者・確認者の署名欠落
・生菌数試験の培養観察記録における観察者署名の欠落
・試験記録のQAレビューにおける照査署名および日付の欠落 |
| 理由 |
ALCOA+の「帰属性」および「同時性」の欠如である。GMP文書において、「誰が・いつ」実施・確認したか不明な記録は無効であり、品質保証の根拠となり得ない。 |
| 事例5 |
管理者不明かつ表示のないファイルや文書が、居室や倉庫等に散乱していた。 |
| 理由 |
文書管理手順からの逸脱である。文書の紛失や、法定保存期間満了前の誤廃棄リスクがある。全ての文書は整理・整頓し、検索可能性(Available)を維持した状態で保管管理されなければならない。 |
| 事例6 |
重要文書(製造記録等)が施錠保管されておらず、持ち出し管理も行われていなかった。実際、貸し出された試験記録が1年以上返却されていない事例があった。 |
| 理由 |
記録の紛失および改ざんリスクに直結する。原本記録は耐火金庫等で施錠管理し、持ち出し・返却の履歴を厳格に管理(ログ管理)する必要がある。 |
| 事例7 |
無菌試験の結果判定等の主観が入りうる試験を、試験者1名のみで実施・判定していた。 |
| 理由 |
写真等の客観的証拠が残らない試験(官能検査、目視検査等)においては、誤判定や恣意的な判断を防ぐため、ダブルチェック(複数名による確認)等の信頼性確保措置が必須である。 |
![]()
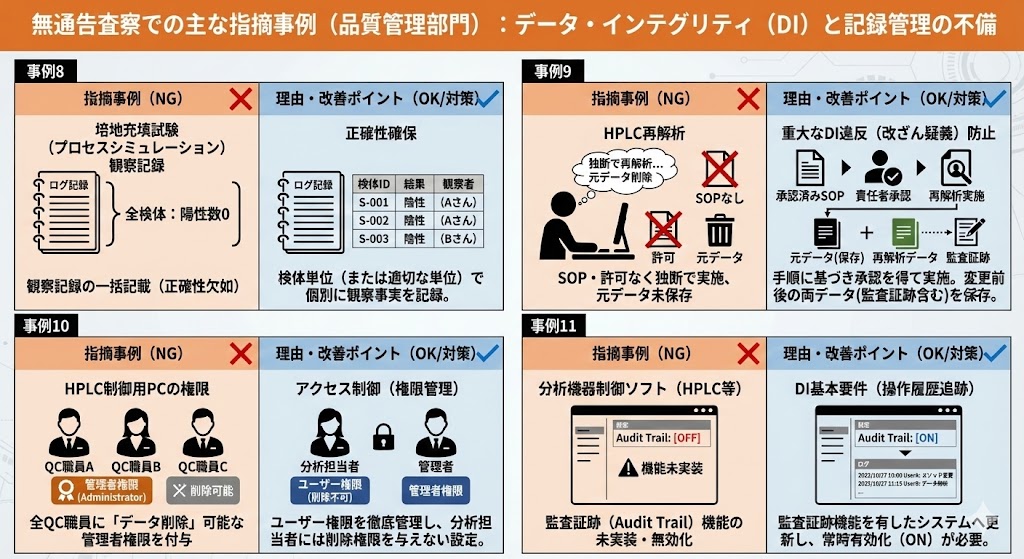
| 事例8 |
培地充填試験(プロセスシミュレーション)の観察記録において、個々の検体ごとではなく、全検体の結果をまとめて「陽性数0」と一括記載していた。 |
| 理由 |
「正確性」の欠如である。観察記録は、検体単位(または適切なトレイ単位等)で識別し、観察者が実際に観察した事実として個別に記録する必要がある。 |
| 事例9 |
HPLC(高速液体クロマトグラフィー)において、以下の状態で頻繁に再解析が行われていた。
・再解析に関するSOP(手順書)が存在しない
・責任者の許可なく担当者が独断で実施
・再解析前の元データが保存されていない
・責任者が再解析の妥当性を確認していない |
| 理由 |
これは典型的なDI違反であり、データの改ざんを疑われる重大な指摘である。再解析は、規定された手順に基づき、正当な理由と責任者の承認がある場合にのみ実施され、かつ変更前後の両データ(監査証跡含む)が保存されなければならない。 |
| 事例10 |
HPLC制御用PCにおいて、全てのQC職員に「データの削除」が可能な管理者権限(Administrator)が付与されていた。 |
| 理由 |
不適切なデータ操作や削除を防止できない。ユーザー権限の管理を徹底し、分析担当者にはデータの削除権限を与えない設定(アクセス制御)を行う必要がある。 |
| 事例11 |
HPLC等の分析機器制御ソフトにおいて、監査証跡(Audit Trail)機能が実装されていない、あるいは機能が無効化されていた。 |
| 理由 |
「誰が、いつ、何をしたか」という操作履歴を追跡できないため、データの信頼性を証明できない。DI対応の基本要件として、監査証跡機能を有したシステムへの更新、および機能の常時有効化(ON)が必要である。 |