
医薬品製造に関わる基礎知識をまとめて紹介します。GMP3つの基本要件とは?GMP3原則とは!?医薬品をつくる工程で大切なことをまとめました。
GMPとは!?
GMPとは…医薬品の製造管理及び品質管理に関する基準で、品質の良い優れた医薬品を製造するための要件をまとめた国際的な指針。

「GMP(ジーエムピー)」という言葉、医薬品業界にいれば毎日のように耳にするはずである。
しかし、マニュアルや法令集の定義は複雑で、本質を掴みにくい。
簡単に言えば、GMPとは「いつ、誰が作っても、必ず安全で効果のある薬ができる仕組み」のことである。
GMPとは何の略か
GMPは、Good Manufacturing Practice の略である。
日本語では「医薬品の製造管理及び品質管理の基準」と訳される。
- Good(良い)
- Manufacturing(製造)
- Practice(実施・習慣)
直訳すれば「良い製造の実践」であるが、これは単なる精神論ではない。
医薬品医療機器等法(薬機法)という法律に基づいた、絶対に守らなければならない法的ルールである。
なぜGMPが必要なのか
もし、あなたがレストランのシェフだと想像してほしい。
「今日は気分が良いから美味しくできた」「昨日は疲れていたから味が薄かった」という料理人に、命を預けられるだろうか。
医薬品は、レストランの料理以上にシビアである。人の命に直結するからである。
しかし、薬には「出来上がった製品をすべて検査することができない」という致命的な特徴がある。
(すべての錠剤を破壊して検査すれば、出荷する製品がなくなってしまうからである)
完成品ですべてを保証できない以上、「作る工程(プロセス)の段階から完璧に管理して、品質を作り込む」必要がある。
これがGMPの基本的な考え方である。
医薬品は、消費者の健康・生命に直接関わるものであるから、その品質の良し悪しはきわめて重要である。そして、どんな偶然の結果によってでも、不良品が消費者のもとに届いてはならない。
体に強く作用する医薬品は、間違いや不正製造は許されないのである。
そこで守るべきルールがGMP(Good Manufacturing Practice:適正製造規範)と呼ばれる製造所における製造管理、品質管理の共通基準である。
GMPは、1968年に世界保健機関(WHO)がその制定を決議し、それを受けて各国で制定されている。
次にGMPのポイントを2つ挙げる。
GMPを守らなければ製造販売できない
医薬品製造では、各々の製造所が自社基準でどんなに厳しく最終試験・検査をしても、医薬品の製造、販売はできない。

GMPは、薬機法に基づき「医薬品及び医薬部外品の製造管理及び品質管理に関する基準」として定められ、この基準に適合しなければ医薬品を製造販売することはできない。
日本で医薬品を市場へ出荷すること、すなわち、製造販売することは医薬品医療機器等法(薬機法)で規制されており、厚生労働大臣の許可・承認を得る必要がある。
医薬品製造業者は、GMPの規制要件に従い、製品の品質と安全性を確保するための品質管理システムを実装し、適切な製造施設や設備を維持することが求められる。
規制当局は、製造業者がGMPを遵守しているかどうかを監査や審査を通じて定期的に評価し、違反があれば適切な対策を取ることがある。
すべての過程における管理基準
GMPは、製品が安全に作られ「一定の品質」が保たれるよう、製造プロセスのすべての段階(原材料の調達から製品の出荷まで)をカバーし、人為的ミスを最小限に抑え、汚染や品質劣化を防ぐことを目指している。
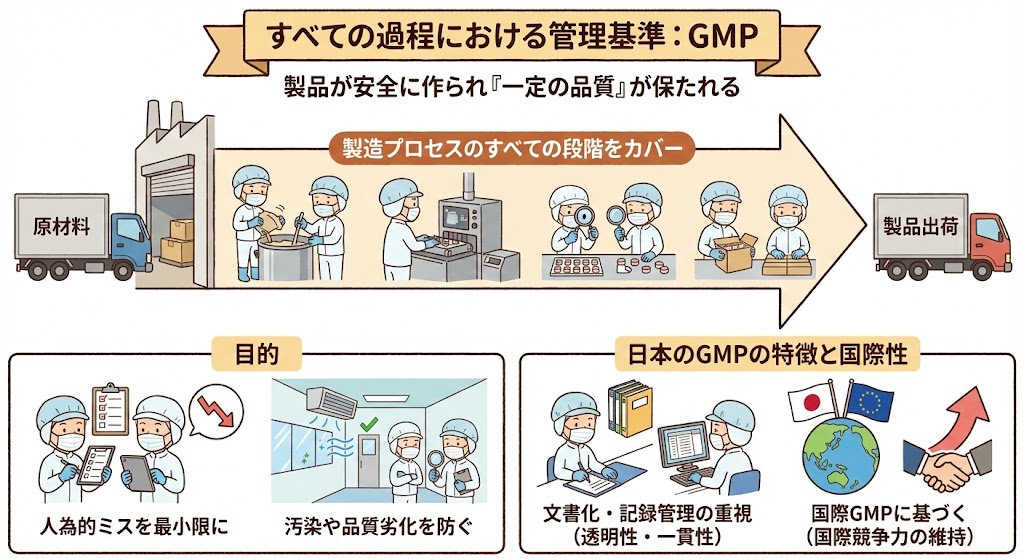
日本のGMPでは、製造業者が製品の品質管理に関するすべての活動を文書化し、記録を適切に管理することが重視されている。これにより、製造プロセスの透明性が確保され、品質管理の一貫性が維持される。
日本のGMPは、国際的な規格である国際GMPに基づいており、日本の製薬企業が国際市場での競争力を維持するための重要な要素となっている。
まとめ
GMPは厳しいルールのように感じるかもしれない。
しかし、これは「この薬なら安心して飲める」という患者からの信頼を守るための、製造者の誇りでもある。
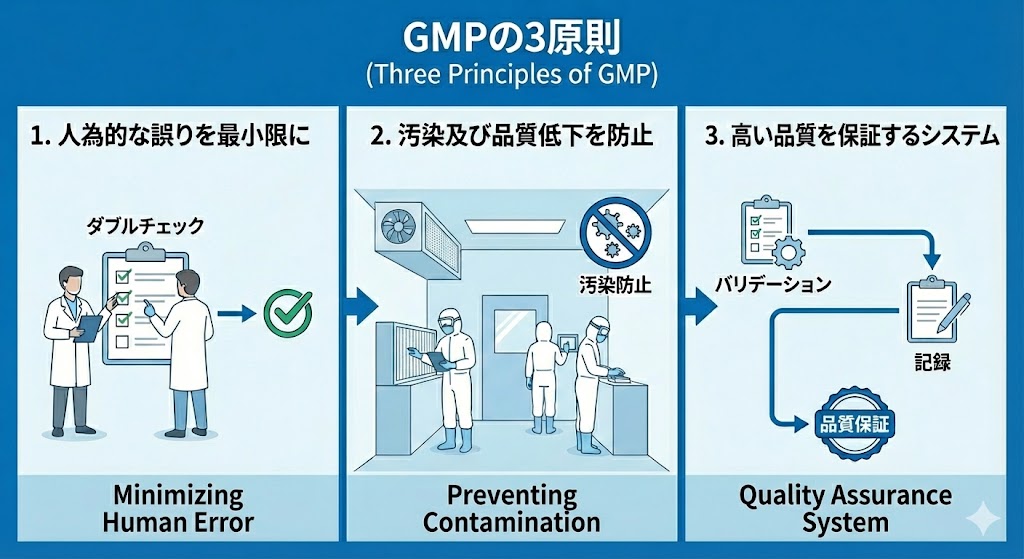
では、具体的にどのような原則に基づいてGMPは運用されているのか。
次ページでは、GMPを理解する上で最も重要な「3つの原則」について詳しく解説する。