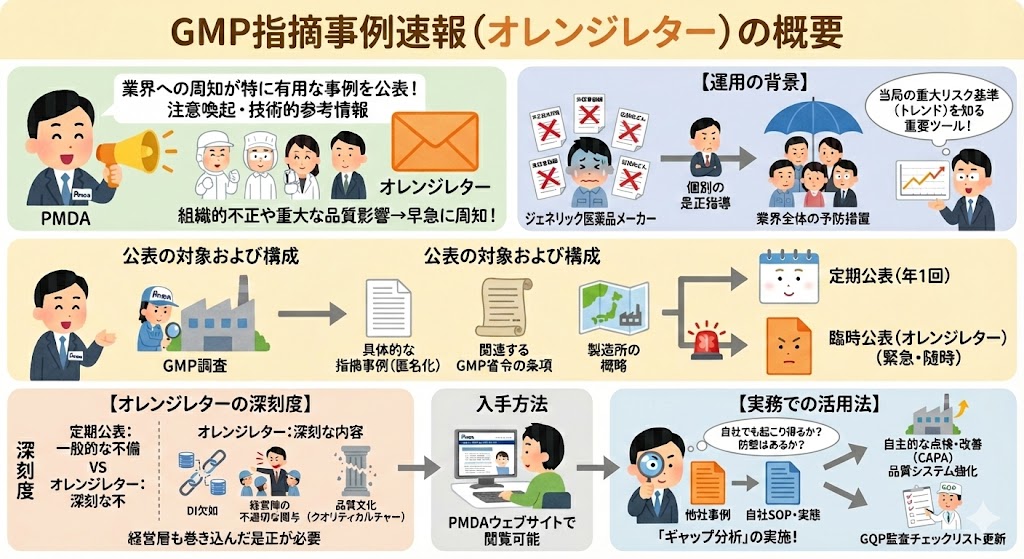
医薬品製造に関わる基礎知識をまとめて紹介します。GMPとは!?医薬品をつくる工程で大切なことをまとめました。
逸脱(いつだつ)とは!?
逸脱(いつだつ)とは!?
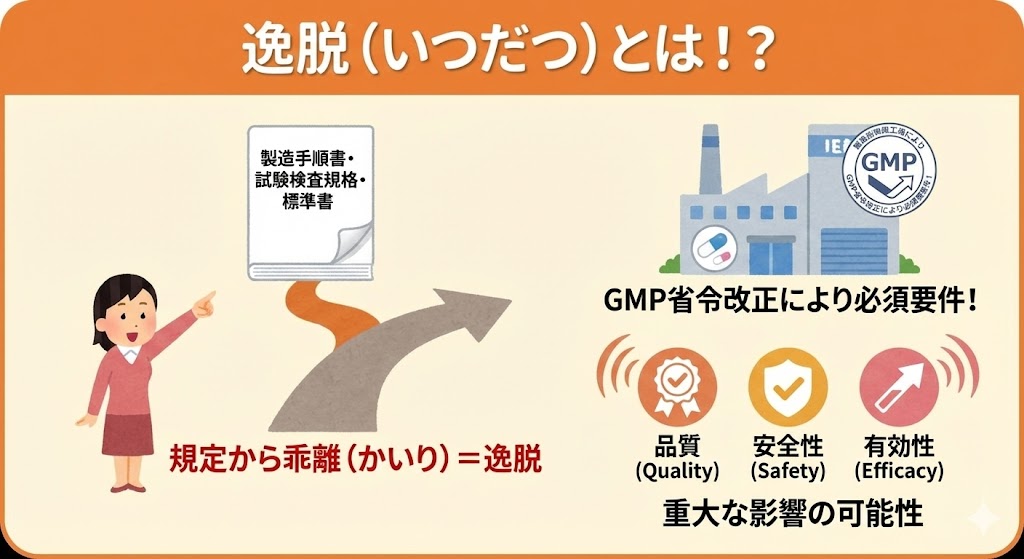
逸脱(いつだつ:Deviation)とは…製造指図書、標準作業手順書(SOP)、試験検査規格、あるいは関連する標準書等の規定された要件から乖離(かいり)している、あらゆる計画外の状況または状態のこと
逸脱は、製造工程や試験検査において発生する「計画外の事象」を指す。これは単なる手順ミスにとどまらず、製品の品質(Quality)、安全性(Safety)、有効性(Efficacy)に重大な影響を及ぼし、ひいてはバリデーション状態(保証された状態)を損なう可能性がある。
そのため、発生した事実を隠蔽・看過することなく、品質リスクマネジメント(QRM)の原則に基づき、その影響を科学的に評価し、厳格な管理と迅速な是正措置を講じることが求められる。
2005年の改正GMP省令施行により制度化され、さらに2021年の改正(PIC/S GMPガイドラインとの整合化)を経て、逸脱管理は医薬品品質システム(PQS)の中核をなす必須要件として位置づけられた。
【逸脱管理の範囲】
逸脱には、「製造パラメータの異常(温度、時間など)」や「手順の不遵守」だけでなく、「資材の破損」や「環境モニタリングの警報レベル超過」なども含まれる。重要なのは、「あらかじめ定められた基準(Standard)」と「実際の結果(Result)」のギャップを全て捕捉するという姿勢である。
また、意図的に手順を一時変更する場合は「逸脱」ではなく「変更管理(Temporary Change)」として扱う点も区別が必要である。
逸脱の発生原因分類
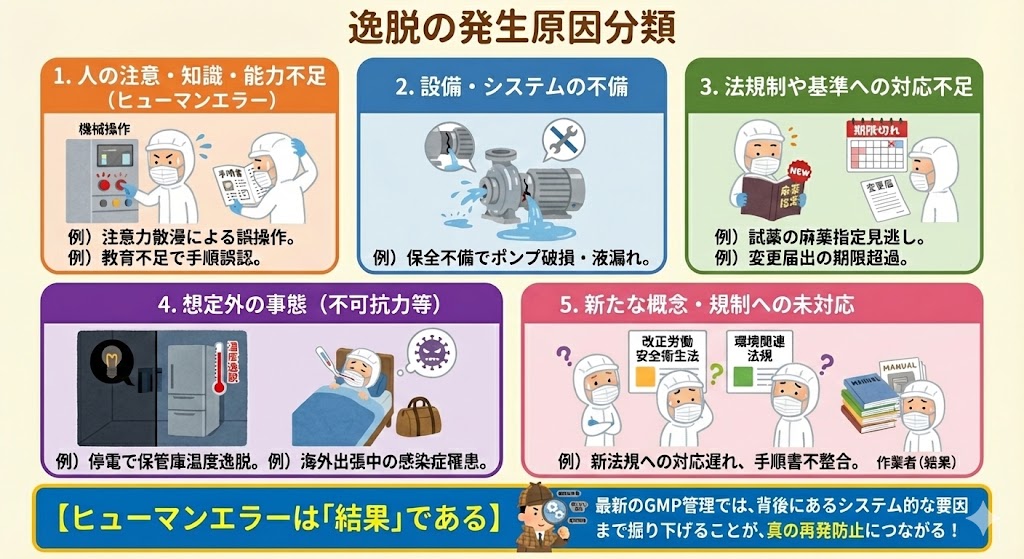
逸脱の発生原因は、主に以下の5つに大別される。
1、人の注意・知識・能力不足(ヒューマンエラー)
例)注意力散漫による機械操作ボタンの押し間違い。
例)教育訓練不足により、バルブ開閉順序を誤認した。
2、設備・システムの不備
例)保全計画の不備により、ポンプのメカニカルシールが破損し液漏れが発生した。
3、法規制や基準への対応不足
例)購入試薬が新たに麻薬指定されたことに気づかず、管理不備として法令違反を問われた。
例)外国製造業者の責任者変更を知らず、変更届出の期限を超過した。
4、想定外の事態(不可抗力等)
例)停電時に自家発電が作動せず、保管庫の温度逸脱が発生し製品廃棄となった。
例)作業者が海外出張中に感染症に罹患し、帰国後の作業従事により汚染リスクが生じた。
5、新たな概念・規制への未対応
例)改正された労働安全衛生法や環境関連法規への対応が遅れ、手順書との整合性が取れなくなった。
【ヒューマンエラーは「結果」である】
最新のGMP管理においては、「作業者の不注意」を根本原因(Root Cause)とすることは推奨されない。なぜその作業者が間違えたのか?「間違えやすい表示だった」「手順が複雑すぎた」「疲労しやすい環境だった」といった背後にあるシステム的な要因まで掘り下げることが、真の再発防止につながる。
GMP省令における規定
GMP省令第15条では、逸脱管理について以下のように規定されている。要約すれば「記録」「評価」「報告」「承認」のプロセス遵守である。
【GMP省令 第十五条(逸脱の管理)】
第十五条 製造業者等は、製造手順等からの逸脱(以下単に「逸脱」という。)が生じた場合においては、あらかじめ指定した者に、手順書等に基づき、次に掲げる業務を行わせなければならない。
一 逸脱の内容を記録すること。
二 重大な逸脱が生じた場合においては、次に掲げる業務を行うこと。
イ 逸脱による製品の品質への影響を評価し、所要の措置を採ること。
ロ イに規定する評価の結果及び措置について記録を作成し、保管するとともに、品質部門に対して文書により報告すること。
ハ ロの規定により報告された評価の結果及び措置について、品質部門の確認を受けること。
2 製造業者等は、品質部門に、手順書等に基づき、前項第二号ハにより確認した記録を作成させ、保管させるとともに、同号ロの記録とともに、製造管理者に対して文書により適切に報告させなければならない。
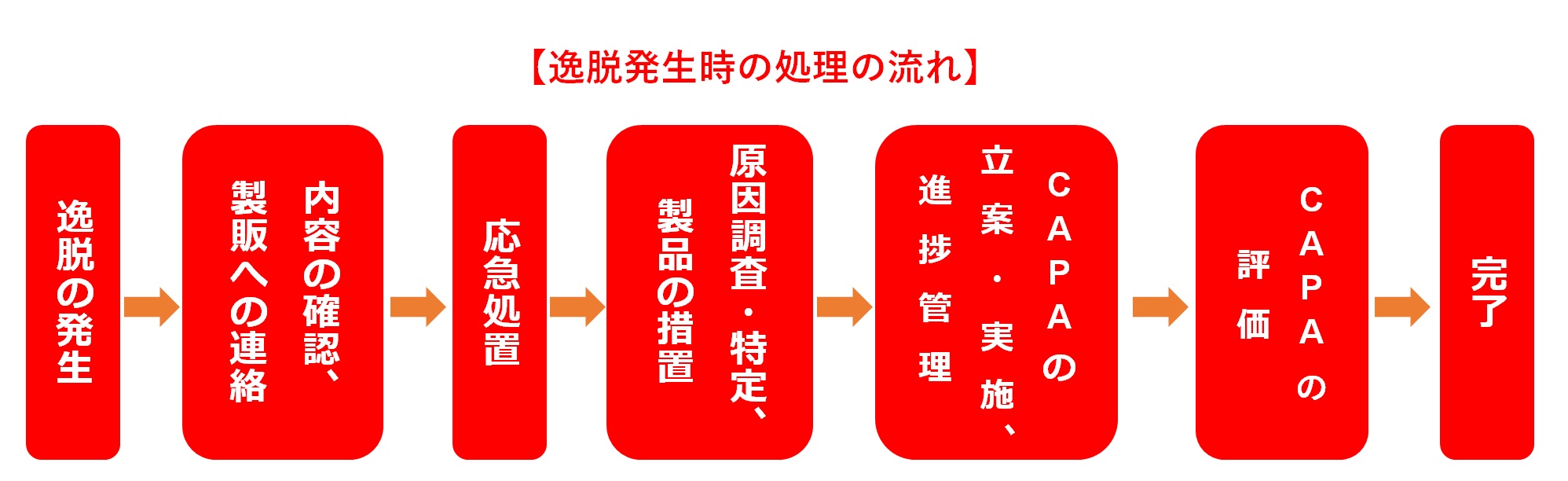
逸脱管理の真の目的とは!?
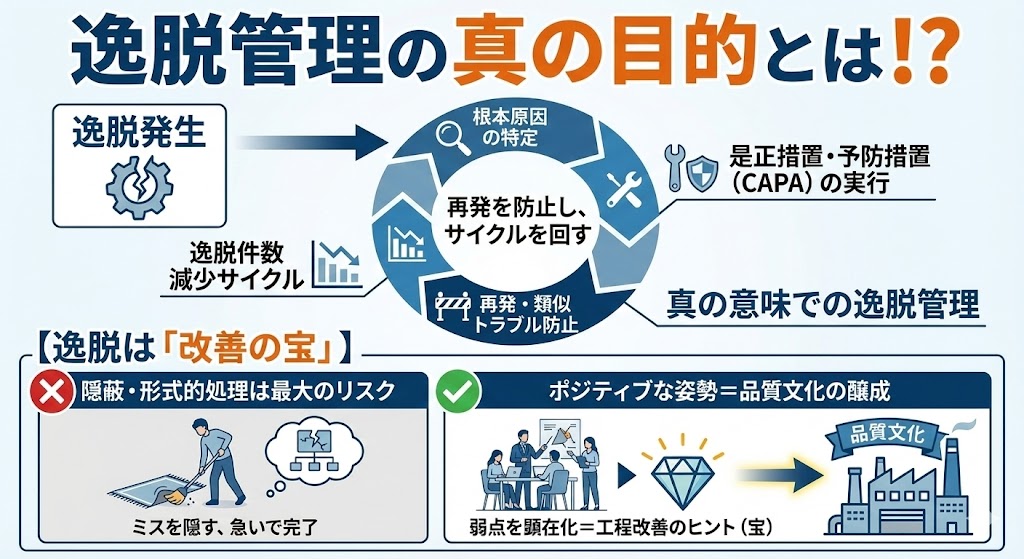
逸脱管理とは、単に記録を残し、その場の処置を行うことだけで完結するものではない。
真の目的は、根本原因を特定し、是正措置・予防措置(CAPA:Corrective Action and Preventive Action)を確実に実行することで、同じ逸脱の再発を防ぎ、類似のトラブルを未然に防止することにある。
つまり、「再発を防止し、逸脱件数を減少させるサイクル」が回っていなければ、真の意味で逸脱を管理しているとは言えないのである。
【逸脱は「改善の宝」】
逸脱を単なる「ミス」として隠蔽したり、処理を急いで形式的に完了させたりすることは最大のリスクである。逸脱は、現在の製造プロセスやシステムに潜む「弱点」が顕在化したものであり、工程改善のヒント(宝)であると捉えるポジティブな姿勢が、品質文化の醸成には不可欠である。
GMP省令(原文)はこちら