
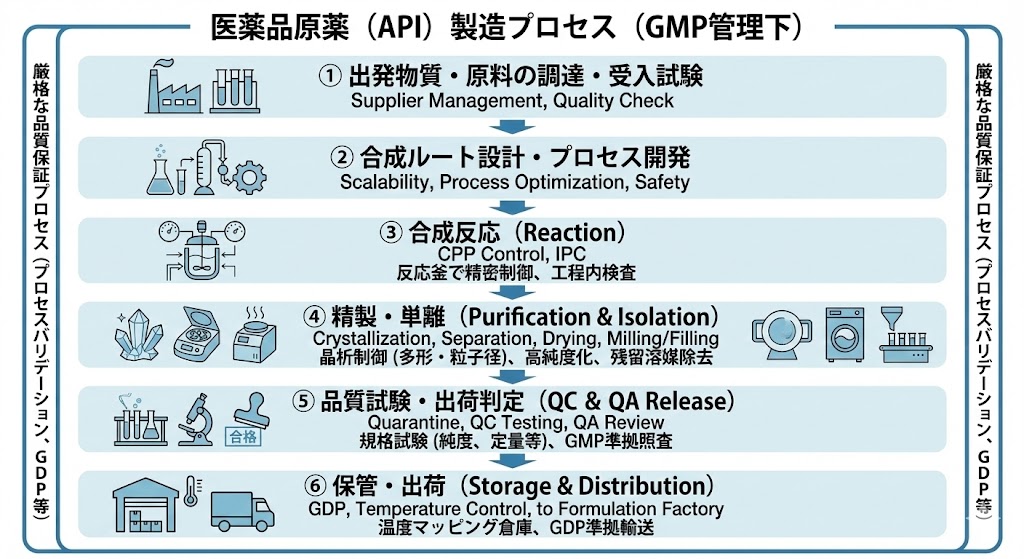
���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���i�����i����j�Ƃ́H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
���i�J���̗���
�@�V��Ì���ɓ͂��܂łɂ́A�����N���Ɩc��Ȕ�p�A�����Ċ����̃n�[�h�������z����K�v������B���i�J���̃v���Z�X�́A���i�ȋK���iGxP�j�̉��Ői�߂���Ȋw�I���̐ςݏd�˂ł���B
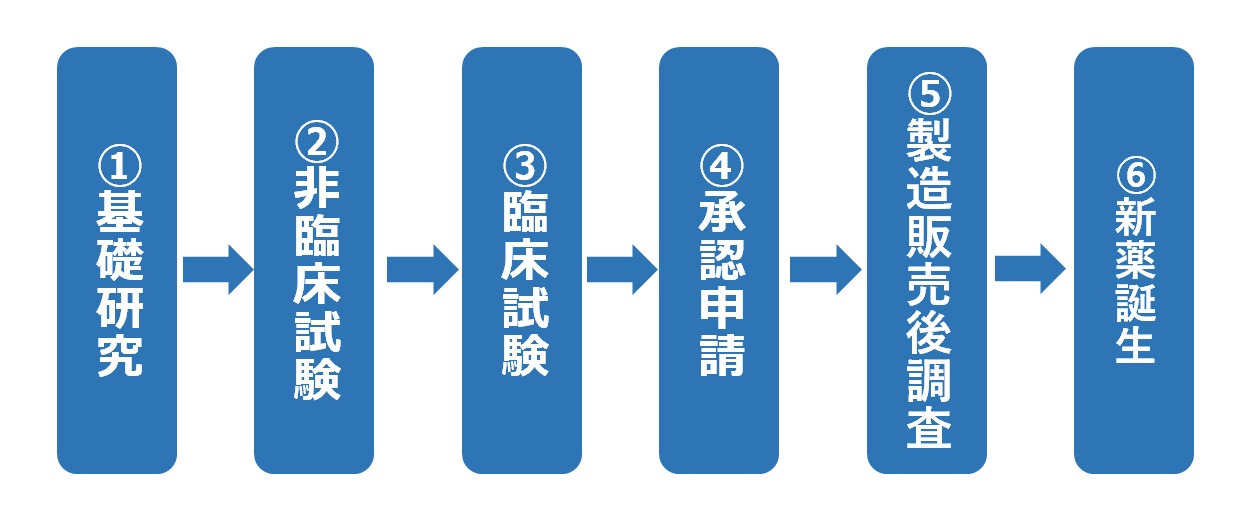
��b�����i�n��^�[�Q�b�g�̒T���E�X�N���[�j���O�j
�@�J���̒[���́A�����̃��J�j�Y���Ɋ�Â����u�n��V�[�Y�i��j�v�̒T���ł���B�����`���\���̉������̒�����A���i�̌��ƂȂ镨����I������B
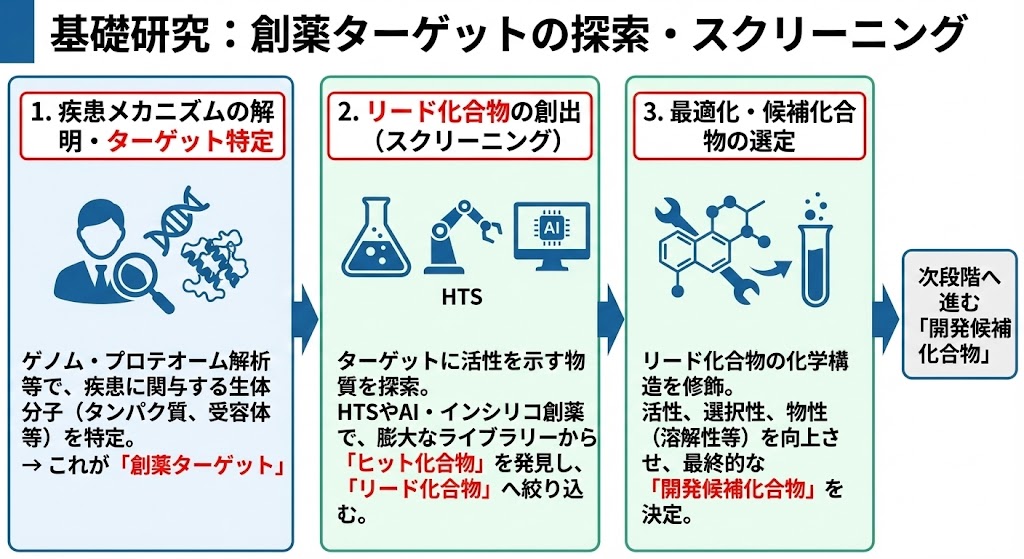
�@�@�������J�j�Y���̉𖾂ƃ^�[�Q�b�g���� �@�Q�m����͂�v���e�I�[����͓�����g���A�����̔��ǂ�i�s�Ɋ֗^���鐶�̓��̕��q�i�^���p�N���A��e�́A�y�f�A��`�q���j����肷��B���ꂪ�n��̕W�I�i�^�[�Q�b�g�j�ƂȂ�B
�@�A���[�h�������̑n�o�i�X�N���[�j���O�j �@���肳�ꂽ�^�[�Q�b�g�ɑ��Ċ���������������T������B�n�C�X���[�v�b�g�X�N���[�j���O�iHTS�j��AI��p�����C���V���R�n��i�R���s���[�^�[�V�~�����[�V�����j����g���A�c��ȉ��������C�u�����[����u�q�b�g�������v�������o���A����ɍœK�����āu���[�h�������v�ւƍi�荞�ށB
�@�B�œK���ƌ�≻�����̑I�� �@���[�h�������̉��w�\�����C�����A�����̋����A�I�𐫁A�����i�n�𐫓��j�����コ����B�ŏI�I�ɁA���i�K�i�ނׂ��u�J����≻�����v�����肷��B
��Տ������iPre-clinical Study�j
�@�q�g�ɓ��^����O�ɁA������|�{�זE��p���ėL�����ƈ��S�����Ȋw�I�ɕ]������v���Z�X�ł���B���̒i�K�̈��S�������́AGLP�iGood Laboratory Practice�F���i�̈��S���Ɋւ����Տ������̎��{�̊�j�ɏ������Č��i�ɍs����B
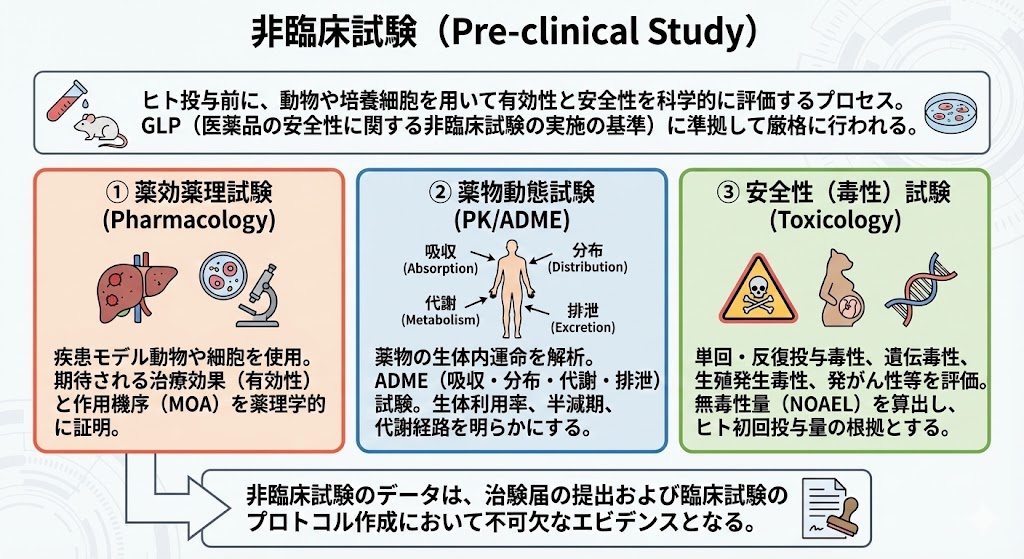
�@�@��������iPharmacology�j �@�������f��������זE��p���A���҂���鎡�Ì��ʁi�L�����j����т��̍�p�@���iMOA�j��w�I�ɏؖ�����B
�@�A���Ԏ����iPK/ADME�j �@�̐��̓��^������͂���B�z���iAbsorption�j�A���z�iDistribution�j�A��ӁiMetabolism�j�A�r���iExcretion�j�̓����������ADME�i�A�h���j�����Ƃ��Ă��B���̗��p���┼�����A��ӌo�H�𖾂炩�ɂ���B
�@�B���S���i�Ő��j�����iToxicology�j �@�P��є������^�Ő��A��`�Ő��A���B�����Ő��A�����Ȃǂ�]������B�ǂ̒��x�̗ʂ܂łȂ���S���i���Ő��ʁFNOAEL�j���Z�o���A�q�g�ł̏��^�ʂ�ݒ肷�鍪���Ƃ���B
�@��Տ������̃f�[�^�́A�����͂̒�o����їՏ������̃v���g�R���쐬�ɂ����ĕs���ȃG�r�f���X�ƂȂ�B
�Տ������i�����FClinical Trial�j
�@�q�g�i���N���l����ъ��ҁj��ΏۂɁA�L�����ƈ��S����������ŏd�v�v���Z�X�ł���BGCP�iGood Clinical Practice�F���i�̗Տ������̎��{�̊�j�Ɋ�Â��A�팱�҂̐l���ی�ƈ��S�m�ۂ��ŗD��Ɏ��{�����B

�@�@��T�������i�t�F�[�Y1�j �@��ɏ����́u���N���l�v��ΏۂƂ���i�R����ܓ��͏����j�B�팱������ʂ���i�K�I�ɑ��ʂ��A���S���i�E�e���j�Ɩ��ԁi�̓��ł̋����j���m�F����B
�@�A��U�������i�t�F�[�Y2�j �@�����́u���ҁv��ΏۂƂ���B�L�����ƈ��S�����m�F����ƂƂ��ɁA�œK�ȗp�@�E�p�ʂ�T������i�O����II���A�����II���j�BPoC�iProof of Concept�F�T�O���j���擾����d�v�Ȓi�K�ł���B
�@�B��V�������i�t�F�[�Y3�j �@�����̊��҂�ΏۂƂ�����K�͂Ȍ��ؓI�����ł���B�������v���Z�{�i�U��j�Ƃ̔�r�����i�����_������d�ӌ��������j���s���A�W�����Âɑ���D�z�����v�w�I�Ɍ�����B
�@�Տ������œ���ꂽ���т��A���F�\���̊j�S�f�[�^�ƂȂ�B�Ȃ��A���F�擾��ɍs���鐻���̔���Տ������́A��W�������Ƃ��Ă��B
���F�\���E�R���iApproval Review�j
�@�Տ������܂ł̑S�f�[�^��CTD�i�R�����E�e�N�j�J���E�h�L�������g�j�Ƃ��Ă܂Ƃ߁A�����J���Ȃɐ����̔����F�\�����s���B�����I�ȐR����PMDA�i�Ɨ��s���@�l���i��Ë@�푍���@�\�j���S���B

�@�@�R���iReview�j �@PMDA�̐R���`�[���ɂ��A�i���iCMC�j�A�A�Ő��A�Տ��̊e�f�[�^�ɂ��āA�Ȋw�I�Ó����ƐM���������������B�u���X�N�E�x�l�t�B�b�g�o�����X�v�̊ϓ_����A���F�̉ۂ����f�����B
�@�A���n�����iGCP/GMP Inspection�j �@�\�������i�f�[�^�j�̐M�������m�F����K�������ʒ�����AGCP���n�������s����B�܂��A�������ɑ��ẮAGMP�iGood Manufacturing Practice�j�K�������������{����A�K�Ȑ����Ǘ��E�i���Ǘ��̐��ɂ��邩���m�F�����B
�@�B�E�H�i�q���R�c��ւ̎���E���\ �@PMDA�̐R�����Ɋ�Â��A�����J���Ȃ̐R�c��Ő��Ƃɂ��R�c���s����B
�@�C���F�iApproval�j �@�����J����b�ɂ�萻���̔����F���t�^�����B����ɂ��A���i�̎s�ꗬ�ʂ��\�ƂȂ�B
�����̔��㒲���iPMS�FPost Marketing Surveillance�j
�@�����̓S�[���ł͂Ȃ��A���Տ��ł̕]���̃X�^�[�g�ł���B�����ł͓����Ȃ������������W���邽�߁AGPSP�i�����̔���̒����y�ю����̎��{�̊�j�����GVP�i�����̔�����S�Ǘ��̊�j�Ɋ�Â��A�p���I�ȊĎ����s����B

�@�@�g�p���ђ����E����g�p���ђ��� �@����f�É��ł̎g�p���ԁA�����⍂��ҁA�����ǂ�L���銳�҂ȂǁA�����ł͏��O����Ă������ґw�ɂ�������S���E�L�������m�F����B
�@�A�s�̒��㒲�� �@�V����̔��N�ԁA�W���I�ɕ���p�������W���A�d�Ăȕ���p�̑��������ƓK���g�p�̊��N���s���B
�@�BRMP�i���i���X�N�Ǘ��v��j�̎��H �@�J���i�K���画�����Ă��郊�X�N�i���肳�ꂽ���X�N�j��A��s�����Ă���_�i�s�����j�ɂ��āA�s�̌�̈��S����v��E���{�E�]������T�C�N�����B
�@�C�ĐR�����x �@�V��͌����Ƃ���8�N�ԁi���a�p���i����10�N�j�A�ĐR�����Ԃ��݂�����B���̊��Ԓ��Ɏ��W���ꂽ�f�[�^����ɁA�L�����ƈ��S���̍Ċm�F���s����B
�V��J���̌���Ɖۑ�
�@��b���������s�Ɏ���m���́A��ʓI��1������1����3������1���x�ƌ�����B�J�����Ԃ�9�`17�N�A�J����p�͐��S���`��牭�~�ȏ��v����n�C���X�N�E�n�C���^�[���ȃv���W�F�N�g�ł���B
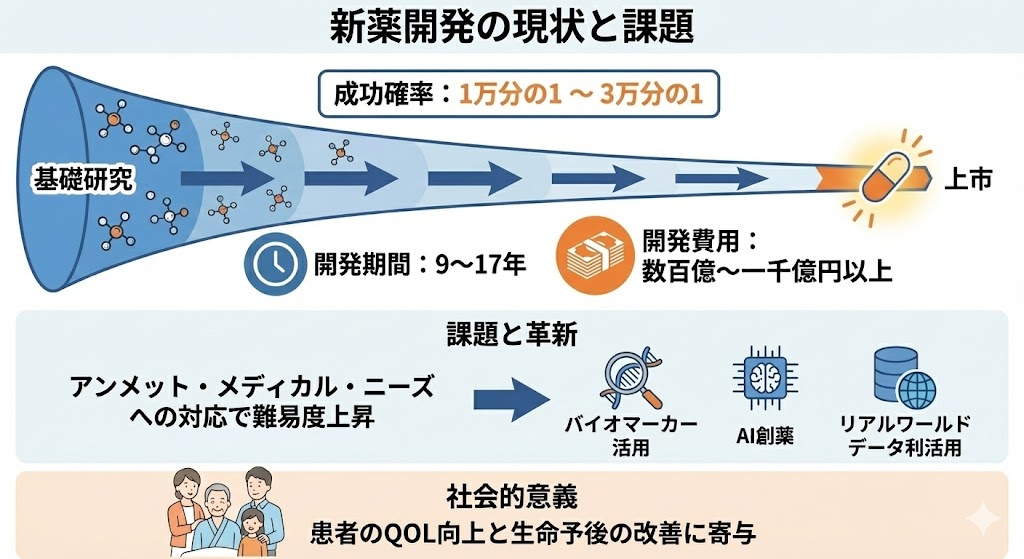
�@�ߔN�́u�A�����b�g�E���f�B�J���E�j�[�Y�i������������Ă��Ȃ���Ãj�[�Y�j�v�ւ̑Ή������߂��A�n��̓�Փx�͔N�X���܂��Ă���B�����m�������コ���邽�߁A�o�C�I�}�[�J�[�̊��p�AAI�n��A���A�����[���h�f�[�^�̗����p�ȂǁA�J���v���Z�X�̊v�V���i�߂��Ă���B
�@�V��̒a���́A���E���̊��҂�QOL�i�����̎��j����Ɛ����\��̉��P�Ɋ�^����A�ɂ߂ĎЉ�I�Ӌ`�̑傫�����ʂł���B
��Ȑ��p����
| GxP (Good x Practice) |
���i�̌����J�����琻���A�̔��A�s�̌���S��Ɏ���܂ł̑S���C�t�T�C�N���ɂ����āA���炷�ׂ��u�K����v�̑��́B x�ɂ͊e�i�K�̃A���t�@�x�b�g������i��FGLP, GCP, GMP, GQP, GVP, GPSP�j�B���S���ƐM������S�ۂ��邽�߂̋K���v���ł���B |
|---|---|
| CMC (Chemistry, Manufacturing and Control) |
����E���܂́u���w�I�����v�u�����H���v�u�i���Ǘ��v�u���萫�v���Ɋւ��錤������уf�[�^�̑��́B �Տ��������u�q�g�ł̗L�����E���S���v������̂ɑ��ACMC�́u���m�Ƃ��Ă̕i���E�P�퐫�v��ۏ����ՂƂȂ�B |
| CTD (Common Technical Document) |
���ĉ��iICH�j�ō��ӂ��ꂽ�A���i���F�\���̂��߂̍��ۋ��ʉ������i�R�����E�e�N�j�J���E�h�L�������g�j�B ���E���ʂ̗l���Ő\���������쐬���邱�ƂŁA�O���[�o���J������ѐR���̌��������}���Ă���B |
| PoC (Proof of Concept) |
�u�T�O���v�Ɩ��B �V���╨�����A�q�g�i���ҁj�ɂ����đz��ʂ�̎��Ì��ʂ��������Ƃ��A�Տ������i��ɑ�U�������j�ŏ��߂Ċm�F����i�K���w���BPoC�̎擾�́A�J����Go/No-Go���f�ɂ�����ő�֖̊�ł���B |
| In silico (�C���V���R) |
�u�V���R���i�R���s���[�^�[�`�b�v�j�̒��Łv�Ƃ����Ӗ��̑���B �����ǁiIn vitro�j��́iIn vivo�j��p���������ł͂Ȃ��A�R���s���[�^�[�V�~�����[�V������p���Ė��ݍ�p��Ő���\�������@�B�J�����Ԃ̒Z�k�Ɋ�^����B |
| Unmet Medical Needs (�A�����b�g�E���f�B�J���E�j�[�Y) |
�����L���Ȏ��Ö@���m������Ă��Ȃ��A��������Ȃ���Ãj�[�Y�̂��ƁB ��a�A�����A�F�m�ǂȂǂ��Y�����A�ߔN�̑n��^�[�Q�b�g�̎嗬�ƂȂ��Ă���B |