���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���i�J���̗���Ƃ́H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
�����H���̊Ǘ��ƒ��ӎ���
�{�e�ł́A���i�����ɂ�����H���Ǘ��̏d�v��������ї��ӓ_�ɂ��Ă܂Ƃ߂�B
���ޗ��̎戵��
���i�̌��ޗ��́A�i����S�ۂ��邽�߂ɏ�Ɍ��i�ȏ������Ŏ�舵���K�v������B
���ޗ��ɂ͐��m�Ȑ������A���b�g�ԍ��A�����N�����Ȃǂ��L�ڂ������x����\�t���A���m�Ɏ��ʂ���B����͐������̍����i�~�b�N�X�A�b�v�j��������邽�߂̕K�{�v���ł���B�܂��A�����Ɏg�p���錴�ޗ��́A���Ђ̎�������ɍ��i�����u�i���m�F�ς݂̌��ޗ��v�Ɍ��肷��B
���ʍH���ɂ����闯�ӓ_
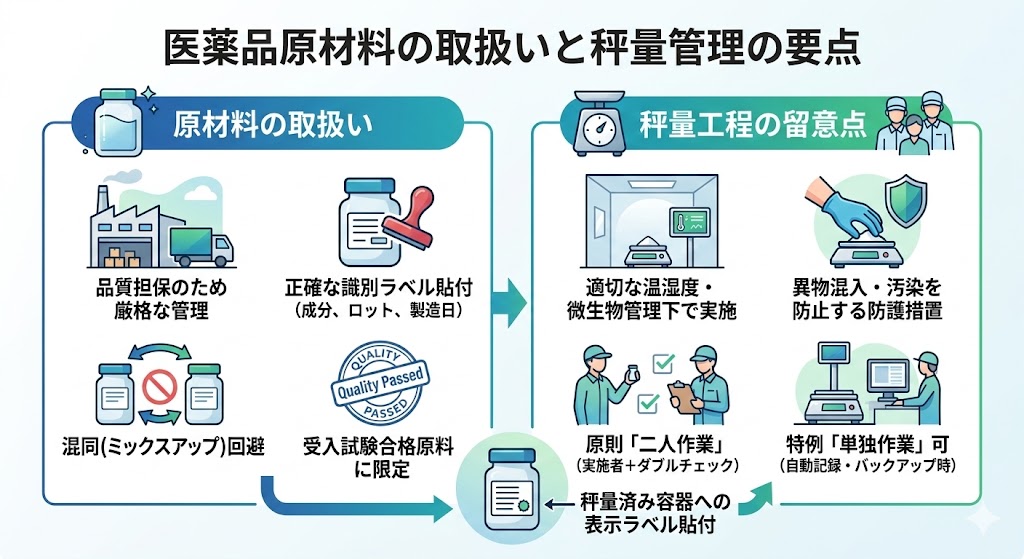
�����̔��ʁE�v�ʎ��ɂ́A�Ώی����Ɉ��e�����y�ڂ��Ȃ��悤�A�K�ȉ��x�E���x�A����є������Ǘ����ō�Ƃ����{����B���킹�āA�ٕ������≘����h�~���邽�߂̊m���Ȗh��[�u���u����B
���ʂ͐��i�i���ɒ�������ԈႢ�̋�����Ȃ��d�v����ł��邽�߁A�����Ƃ��āu��l��Ɓv�Ŏ��{����B
��l�����ۂ̌v�ʂƋL�^���s���A������l�����̋L�^�̐��m�������A���^�C���Ŋm�F�i�_�u���`�F�b�N�j����B�������A�����L�^�v�𓋍ڂ����V�������g�p���A�v�ʌ��ʂ��V�X�e����Ɏ����Ńo�b�N�A�b�v���������ɂ����ẮA�P�Ƃł̔��ʍ�Ƃ����e�����B
���ʁE�v�ʍς݂̌��ޗ������߂��e�e��ɂ́A���e���������u�\�����x���v��K���\�t����B
�����H���ɂ����闯�ӓ_
�e���ޗ��̎g�p���т͌����ɋL�^����A�����L�^����b�g�L�^���ɋL�ڂ����B����ɂ��A���i�̊��S�ȃg���[�T�r���e�B�i�ǐՉ\���j���m�ۂ����B
���i�����ɂ����ẮA�d�v�ȍ�ƍH���ɕK���������z�u����B
�d�v��ƂƂ́A���i�i���ɏd��ȉe�����y�ڂ��\���̂��������w���B��̓I�ɂ́A�����̔��ʁE�v�ʁA���ޗ��̎d���݁A�d�v�H���ɂ�����@�푀��A�ŏI���i�̎�舵���Ȃǂ��Y������B�����̍�Ƃɂ͗���҂�݂���ƂƂ��ɁA�����L�^�����L�^�m�F�җ���݂��A�ӔC�̏��݂m�ɂ���B
���ʁiYield�j�̊Ǘ�
���i�����ɂ�����u���ʁiYield�j�v�Ƃ́A�����v���Z�X�̓���̒i�K�ɂ����ē�����ړI���̗ʂ������p�����[�^�ł���B��̓I�ɂ́A�����������ޗ����ϊ��v���Z�X���o�āA�ŏI�I�ɂǂ̒��x�̗ʂ����i�Ƃ��ĉ�����ꂽ����\���B
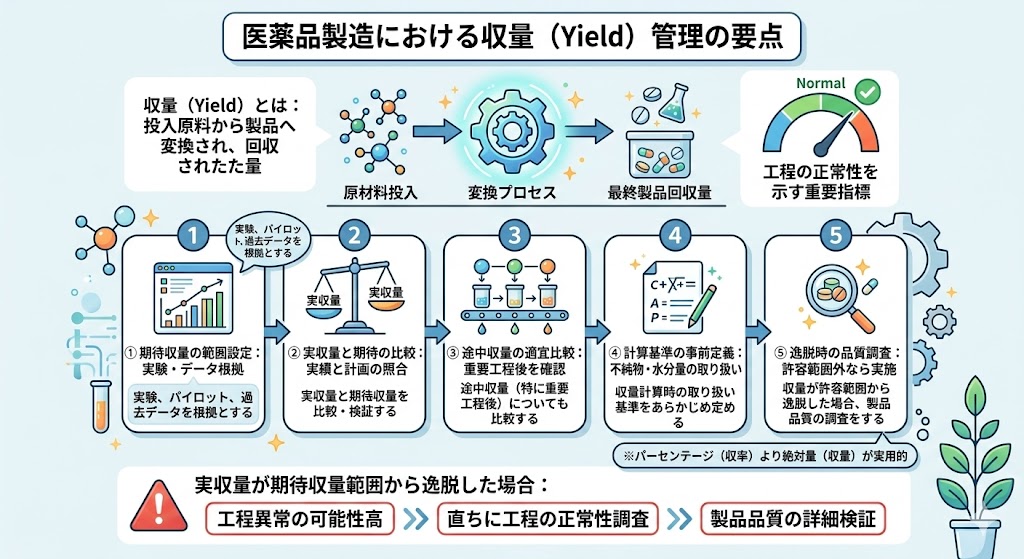
�����́A�Y�����鐻���H��������ɉғ��������������d�v�Ȏw�W��1�ł���B���ʊǗ��̊�{�I�ȗ�����ȉ��Ɏ����B
�����ʊǗ��̗��ꁄ
�@ ���Ҏ��ʂ̋��e�͈͂�ݒ肷��i�����f�[�^�A�p�C���b�g�X�P�[���A�ߋ��̐����f�[�^�������Ƃ���j
�A �����ʂƊ��Ҏ��ʂ��r�E������
�B �r�����ʁi���ɏd�v�H����̎��ʁj�ɂ��Ă��K�X��r����
�C ���ʌv�Z���ɂ�����s�����ʂ␅���ʂ̎�舵��������炩���ߒ�߂Ă���
�D ���ʂ����e�͈͂����E�����ꍇ�́A���i�i���̒��������{����
��������A�p�[�Z���e�[�W�ŕ\���u�����v�����A��Ηʂł���u���ʁv��p��������H����r�ɂ����Ď��p�I�ł���B
�����ʂ����Ҏ��ʂ͈̔͂����E�����ꍇ�A�Y���H���ɉ��炩�ُ̈킪���������\���������B�����ɍH���̐��퐫��������ѐ��i�i���̏ڍׂȌ����s���K�v������B
�H����Ƃ̎��Ԑ���
�e�����H���ɂ́A���m�ȍ�Ǝ��Ԑ����i�^�C�����~�b�g�j��ݒ肷��B���̊���Ԃ́A�p�C���b�g�X�P�[���ł̐����f�[�^��A���Ɛ��Y�ɂ�����ߋ��̎��т����ƂɑÓ�����]�����Č��肷��B
�ݒ肳�ꂽ�������ԓ��ɍ�Ƃ��������Ȃ������ꍇ�́A�@�B�I�g���u����菇�̌��Ȃlj��炩�ُ̈킪���������Ɛ��肳��邽�߁A��E�����Ƃ��đΉ�����B�����ɐ��i�i���ւ̉e�����E�]������ƂƂ��ɁA���{�������������A�K�Ȑ����[�u�iCAPA�j���u����B
�H���������ƃT���v�����O
�H������������уT���v�����O�́A�����v���Z�X�̏�Ԃ����A���^�C���ŊĎ����A�ŏI�I�Ȑ��i�i����S�ۂ��邽�߂ɕs���ȕi���Ǘ���Ƃł���B
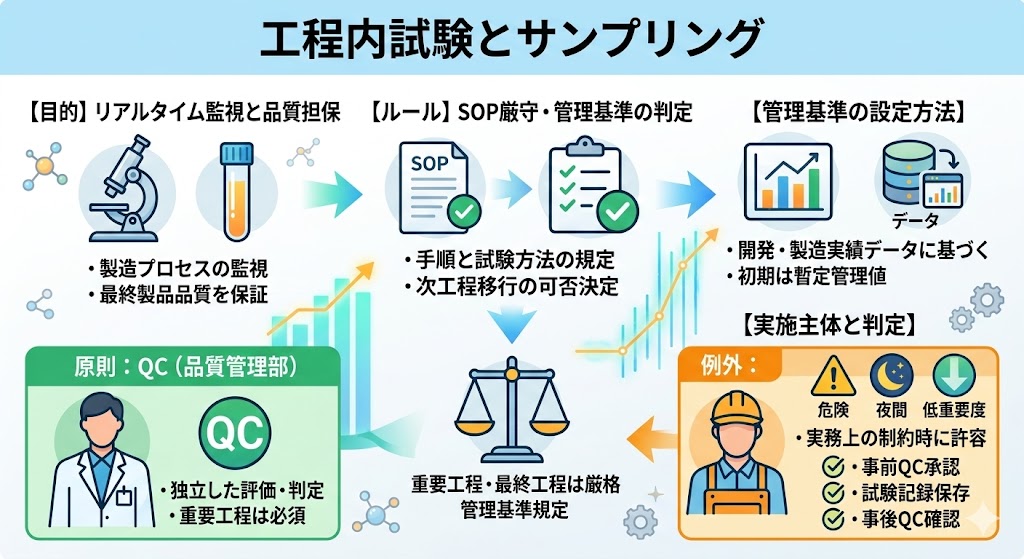
��ʓI�ɁA���i�i���ɒ��ډe����^����H����ΏۂƂ��Ď��{����A�T���v�����O�菇�⎎�����@�̏ڍׂ��߂�SOP�i�W����Ǝ菇���j�����炵�čs����B
�������ʂɊ�Â��H���̈��萫��]�����A���H���ւ̈ڍs�ۂ����肷�邽�߁A�Ǘ���i�K�i�l�j�̓K�Ȑݒ肪�ɂ߂ďd�v�ƂȂ�B���ɏd�v�H����ŏI�H���ɂ����ẮA��茵�i�ȊǗ����݂���B�Ǘ��l�͊J���i�K�̃f�[�^������т��瓱���o����邪�A���������i�K�ł͎b��Ǘ��l��p���ĉ^�p���邱�Ƃ������B
�H�����T���v�����O�A�����̎��{�A����ь��ʂ̔���́A�Ɨ������]����S�ۂ��邽�������Ƃ���QC�i�i���Ǘ����j�����{�������Ƃ��]�܂����B�d�v�H���̊m�F�͕K��QC���S������B
�������A�댯���T���v�����O��ƁA��ԃV�t�g���̑Ή��A�d�v�x�̒Ⴂ�H���������ȂǁA������̐�����ꍇ�́A�����S��������s���邱�Ƃ����e�����B���̏ꍇ�A�T���v�����O����ю������@�ɂ��āA���O��QC����̏��F�Ă����Ȃ���Ȃ�Ȃ��B��ƌ�͂��ׂĂ̎����L�^��ۑ����A�����QC�̊m�F�Ə��F����K�v������B
�H���ɂ����鉘���h�~��
���i�����ɂ����鉘���h�~�i�R���^�~�l�[�V�����E�R���g���[���j�́A���i�̗L�����Ɗ��҂̈��S��������ōŏd�v�ۑ��1�ł���B�H�����̉����h�~�Ɋ֘A�����{����ȉ��Ɏ����B
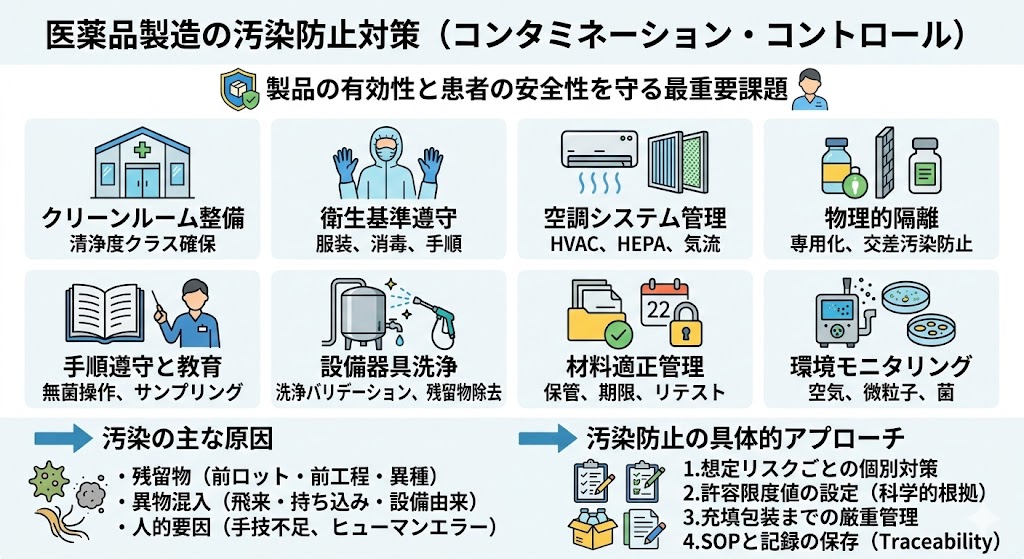
| �N���[�����[������ѐ��̐��� | �����H�����s����G���A�́A��߂�ꂽ����x�N���X�����N���[�����[����Ƃ��Đ����E�Ǘ�����B����ɂ��A�O������̔����q��������̐N�����ŏ����ɗ}������B |
|---|---|
| �q����̌��i�ȏ��� | �S�]�ƈ��́A��߂�ꂽ�q��������炵�č�Ƃɂ�����B����ɂ́A���o�߂�ŋێ�܂̐��������p�A�K�Ȏ�w���ŁA����ѓ��ގ����̉q���菇�̗��s���܂܂��B |
| �E���C�V�X�e���iHVAC�j�̊Ǘ� | �N���[�����[�����̋V�X�e������э����\�t�B���^�[�iHEPA�t�B���^�[���j�́A�����q�E�����������ʓI�ɏ������A������C���𐧌䂷�邽�߂ɒ���I�ȕێ�E�_�����s���ł���B |
| �K�ȕ����I�u�� | �قȂ鐻�i��H���̐����G���A�͕����I�Ɋu�����A��p���܂��͎��ԓI�u�����s���B����ɂ��A�َ퐻�i�Ԃ̌��������i�N���X�R���^�~�l�[�V�����j��h�~����B |
| ��Ǝ菇�̏���Ƌ���P�� | �]�ƈ��ɑ��āA�@��̐�����������@�A���ۓI����A�T���v�����O�菇�ȂǁA�������X�N���ŏ������邽�߂̌p���I�ȋ���P�������{����B |
| �ݔ��E���̐��i�N���[�j���O�j | �g�p����ݔ�����́A�o���f�[�V�����i�Ó����m�F�j���ꂽ�菇�Ɋ�Â��O��I�ɐ��B�O���b�g�̎c���������S�ɏ������A���v���Z�X�ւ̎������݂�h���B |
| �ޗ��̓K���ۊǂƊǗ� | ���ޗ��⒆�Ԑ��i�́A�w�肳�ꂽ���x�E���x�������ŕۊǂ���B�܂��A�g�p������e�X�g���������ɊǗ����A�i���ɂ�鉘�����X�N��r������B |
| �����j�^�����O�̎��{ | �����G���A���̐���x���ێ����邽�߁A��C���̔����q�������A���V�ہE�����ہE�t���ۂȂǂ̒���I�Ȋ����j�^�����O���������{����B |
������A�H�����ʼn��������������ꍇ�A��Ɉȉ��̂悤�Ȍ��������������B
���H���������̎�Ȍ�����
�@ ���ꐻ�i�ɂ�����O���b�g�̎c�����i���s�ǁj
�A ���ꐻ�i�ɂ�����O�H���̎c����
�B �َ퐻�i�i�ߋ��ɐ��������ʂ̈��i�j�̎c����
�C �������s�Ő����E�v�ʂ��Ă���َ함���̍���
�D �O������̔��A�܂��͐l�E���ނɂ�鎝�����݈ٕ�
�E �����ݔ��ɗR������ٕ��i���Օ��A�������A�p�b�L���ЂȂǁj
�F �l�I�v���i�s�K�Ȏ�Z�A������̔��o�A�є��̗����A���̑��̃q���[�}���G���[�j
�����̉�����h�����߁A�ȉ��̑���I�Ɏ��{����B
�������h�~�ւ̋�̓I�ȃA�v���[�`��
�@ �z�肳��鉘�����X�N�i�������E�o�H�j���ƂɌʋ�̓I�Ȗh�~������肷��
�A �Z�p�I�Ɋ��S����������Ȏc�����ɂ��ẮA�Ȋw�I�����Ɋ�Â����e���x�l��ݒ肵�Ǘ�����
�B ���������ォ��[�U�E��Ɏ���܂ł́A���h���ȏ�Ԃ̐��i��舵�����ɂ߂Č��d�ɊǗ�����
�C SOP���ŐV��ԂɈێ����A���ׂĂ̍�ƋL�^���m���ɕۊǂ��邱�ƂŁA�ُ픭�����̐v���Ȍ��������i�g���[�T�r���e�B�j�ɔ�����