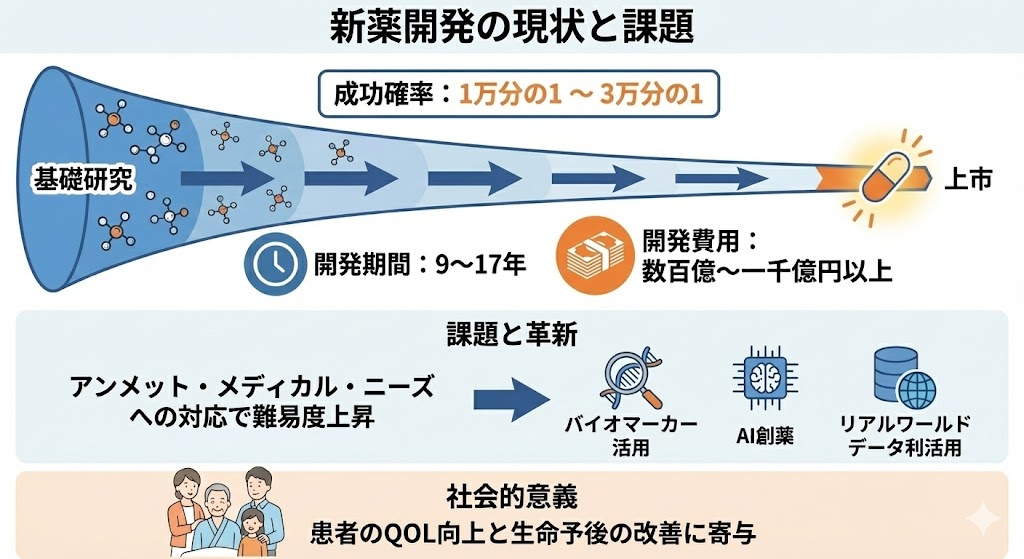
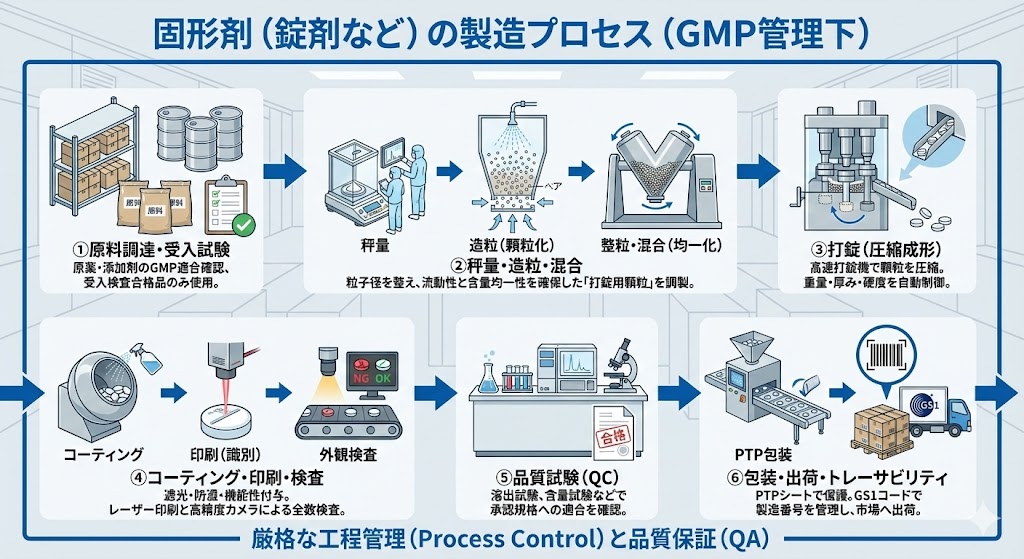
医薬品製造に関わる基礎知識をまとめて紹介します。医薬品開発の流れとは?医薬品をつくる工程で大切なことをまとめました。
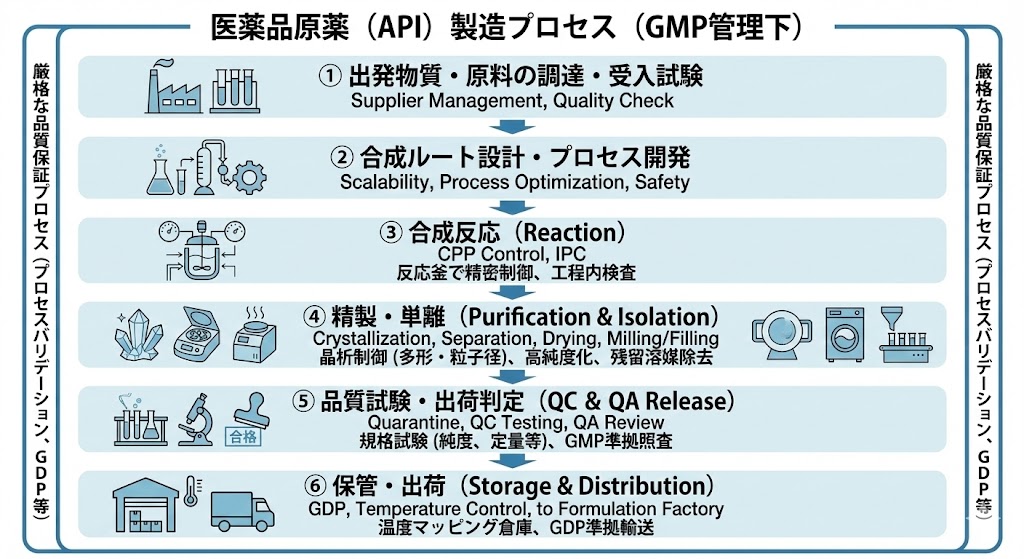
原薬製造の流れ
医薬品の核となる「原薬(API)」の製造は、単なる化学合成ではなく、厳格なGMP(Good Manufacturing Practice)管理下で行われる高度な品質保証プロセスである。
出発物質・原料の調達と受入試験
原薬の品質(CQA:重要品質特性)を確保するためには、その源流となる「出発物質(Starting Material)」および「原材料」の厳格な管理が不可欠である。
ICH Q7(原薬GMPガイドライン)に基づき、サプライヤー選定から受入までの一貫した品質保証体制が敷かれる。
【サプライヤー管理(Supplier Qualification)】
原材料メーカーに対し、品質システムや製造管理状況を確認する「実地監査(Audit)」を行う。
製造方法の変更管理(Change Control)や、不純物プロファイルに関する取り決めを交わし、信頼できる「認定サプライヤー」のみから調達する。
【受入試験と重要素材特性(CMA)】
工場搬入時には、規格適合性だけでなく、原薬の品質に影響を与える「重要素材特性(CMA)」を確認する。
特に、出発物質に含まれる微量な「変異原性不純物」や「異性体」は、後の工程で除去(パージ)できないリスクがあるため、この段階での厳密な制御が求められる。
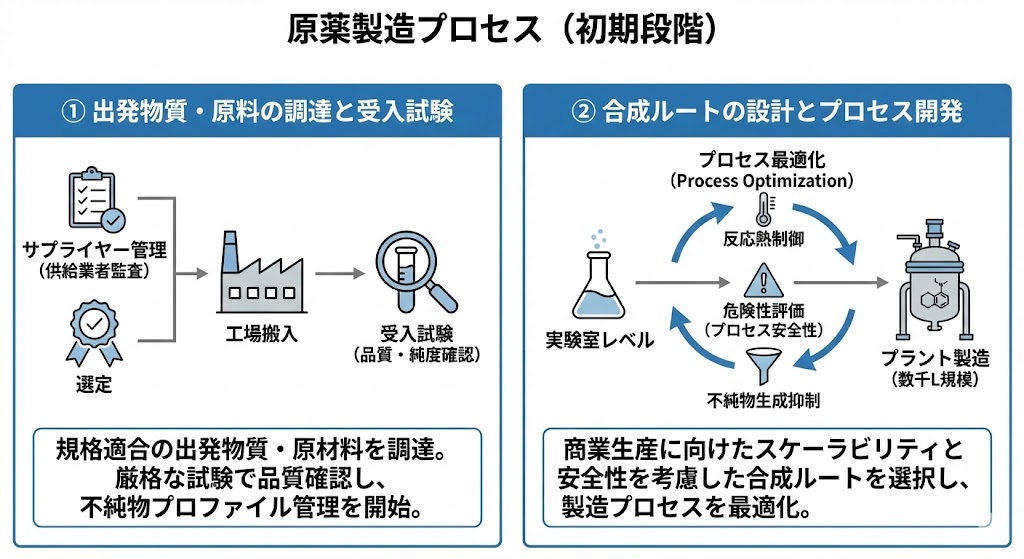
合成ルートの設計とプロセス開発
創薬段階の実験室レベル(メディシナルケミストリー)の製法を、商業生産レベル(プロセスケミストリー)へと最適化するフェーズ。
QbD(Quality by Design)のアプローチを用い、品質を工程の中に作り込む「デザインスペース」の確立を目指す。
【プロセス最適化(Process Optimization)】
実験計画法(DoE)等を活用し、反応温度、時間、pH、撹拌速度などのパラメータが品質に与える影響を解析する。
収率向上だけでなく、「不純物の生成抑制」や「結晶多形の制御」が可能で、かつバラツキに対して堅牢(ロバスト)な製造条件を決定する。
【スケールアップと安全性評価】
フラスコ(数グラム)からプラント釜(数トン)へスケールアップする際、最大の課題となるのが「除熱」と「混合」である。
反応熱測定(DSCやRC1等)による徹底した「プロセス安全性評価」を行い、熱暴走(Runaway Reaction)のリスクがない安全な運転条件を確立する。
合成反応(Reaction)
決定された製造手順に基づき、反応釜(リアクター)にて化学反応を行う。
温度、圧力、撹拌速度、滴下時間などの「重要工程パラメータ(CPP)」を厳密に制御し、反応の進行を管理する。必要に応じ、In-process Control(IPC:工程内検査)を実施し、反応の終点を見極める。
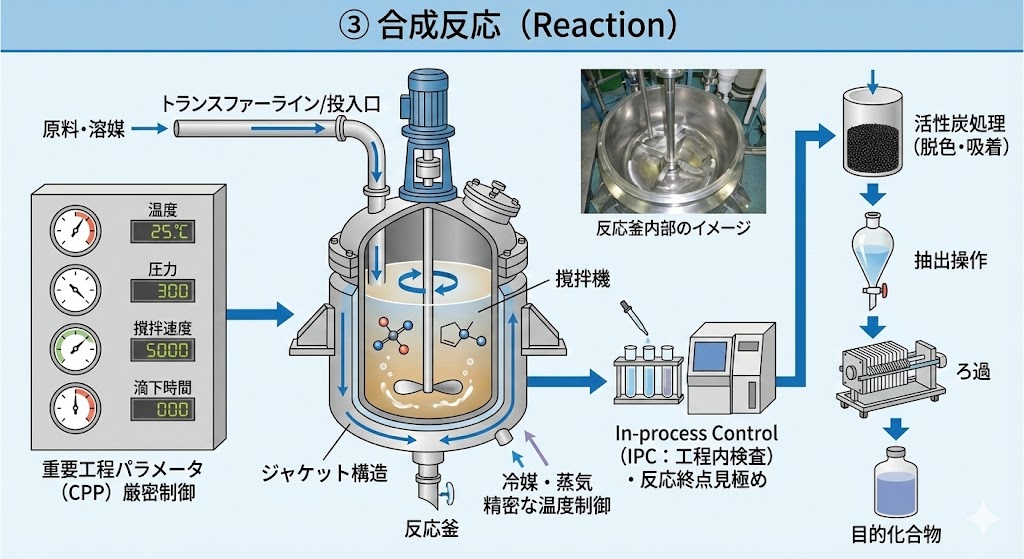
【反応工程の実際】
秤量された原料や溶媒は、配管(トランスファーライン)や投入口を通じて反応釜へ投入される。
反応釜はジャケット構造になっており、冷媒や蒸気を用いて精密な温度制御を行いながら化学反応を進行させる。
反応終了後、目的とする化合物以外の不純物や副生成物を除去するため、活性炭処理(脱色・吸着)や抽出操作、ろ過などを行う場合がある。
精製と単離(Purification & Isolation)
反応混合物から目的物質を高純度に取り出すため、晶析(結晶化)やクロマトグラフィーなどの精製操作を行う。
特に原薬製造においては、最終的な品質(バイオアベイラビリティ等)に直結する「結晶多形」や「粒子径」の制御が極めて重要である。
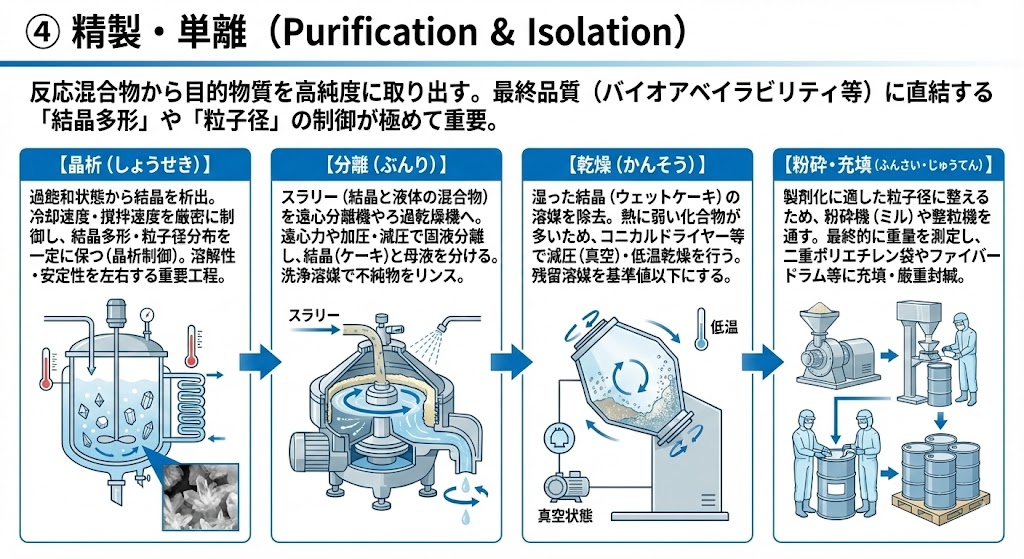
【晶析(しょうせき)】
過飽和状態にした溶液から結晶を析出させる操作。
冷却速度や撹拌速度などの条件を厳密に制御することで、目的とする結晶形(多形)や粒子径分布を一定に保つ(晶析制御)。これは原薬の溶解性や安定性を左右する重要工程である。
【分離(ぶんり)】
スラリー(結晶と液体の混合物)を遠心分離機やろ過乾燥機へ移送する。
高速回転による遠心力や加圧・減圧を利用して、結晶(ケーキ)と母液(不要な液体)を固液分離する。その後、洗浄溶媒を用いて結晶表面の不純物を洗い流す(リンス)。
【乾燥(かんそう)】
湿った結晶(ウェットケーキ)に含まれる溶媒を、コニカルドライヤーなどの乾燥機を用いて除去する。
熱に弱い化合物も多いため、減圧下(真空状態)にて低温で効率的に乾燥させることが一般的である。ここでは残留溶媒が基準値以下になるまで乾燥を行う。
【粉砕・充填(ふんさい・じゅうてん)】
乾燥後の原薬は、製剤化(錠剤などを造ること)に適した粒子径に整えるため、粉砕機(ミル)や整粒機を通すことが多い。
最終的に、重量を測定しながら二重のポリエチレン袋やファイバードラム等の容器に充填し、厳重に封緘(ふうかん)する。
品質試験と出荷判定(Quality Control & Release)
製造された原薬は隔離(Quarantine)され、品質管理部門(QC)による出荷試験を受ける。
「原薬規格」に基づき、確認試験、純度試験(不純物)、定量法、残留溶媒、粒子径などの項目を理化学機器を用いて分析する。
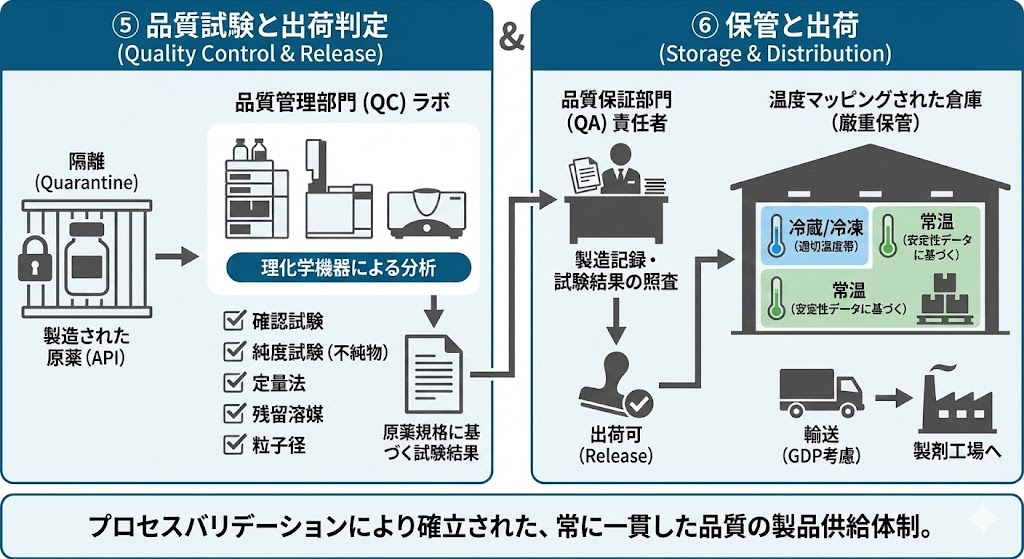
保管と出荷(Storage & Distribution)
品質保証部門(QA)の責任者による製造記録および試験結果の照査を経て、正式に出荷可(Release)となる。
出荷された原薬は、GDP(適正流通基準)を考慮し、温度マッピングされた倉庫で厳重に保管される。製品の安定性データに基づき、常温、冷蔵、冷凍など適切な温度帯で管理され、製剤工場へと輸送される。
原薬製造プロセスは、科学的根拠に基づくプロセスバリデーションによって確立されており、常に一貫した品質の製品を供給できるよう管理されている。
原薬製造に関する主な用語解説
| API (Active Pharmaceutical Ingredient) |
「医薬品有効成分」のこと。いわゆる「原薬」であり、生体に対して薬理作用(治療効果)を発揮する物質そのものを指す。 |
|---|---|
| CPP (Critical Process Parameter) |
「重要工程パラメータ」。 温度、pH、撹拌速度、反応時間など、変動すると製品の品質(CQA)に重大な影響を及ぼすため、厳密に管理・監視しなければならない製造条件のこと。 |
| IPC (In-Process Control) |
「工程内検査」。 製造工程の途中で行われる試験やチェックのこと。反応が完了したか、不純物が除去できているか等を次の工程に進む前に確認し、品質を作り込むために実施される。 |
| 結晶多形 (Polymorphism) |
同じ化学式(成分)であっても、結晶の並び方(構造)が異なる状態。 多形が異なると「溶けやすさ」や「安定性」が変化し、薬の効き目(バイオアベイラビリティ)に影響するため、原薬製造では特定の結晶形を作り分ける技術が必須となる。 |
| Quarantine (クアランティン/隔離) |
試験結果が出るまでの間、製品を使用・出荷できないように物理的またはシステム的に隔離されている状態。「試験中」であることを明確にし、未承認の製品が誤って市場に出ることを防ぐ。 |
| プロセスバリデーション (Process Validation) |
設定された製造手順に従えば、「いつ誰が作っても、規格に適合した製品が恒常的に製造できる」ことを科学的に検証し、文書化すること。商業生産を開始する前の必須要件である。 |