�i���Ɋւ���K�C�h���C���uICH Q1�`Q14�v�Ƃ́I�H
�@ICH�ł́AISO�̍l�������x�[�X�Ɉ��i�ƊE�̗v�f�������ꂽ�i���V�X�e���̃K�C�h���C�����쐬���A���\���Ă���B
�@�i���Ɋւ���K�C�h���C���́A2020�N���_��ICH Q1�`Q14�܂ő��݂��Ă���B
�@

�@�����J���Ȃ���ʒm������i�X�e�b�v5�j�܂ŒB���Ă���̂́A2020�N���_��ICH Q1�`Q11�ł���B
�@���ɁA��ȓ��e�������B
| �K�C�h���C�� | �K�C�h���C���̎�ȓ��e | �ʒm���E ���������N |
|---|---|---|
Q1 |
�y���萫�z
��Q1A�iR2�j�u���萫�����K�C�h���C���v��
�E�h�b�g���萫�K�C�h���C���̉���łł���A�d�b�A���{�y�ѕč�3�ɓ��ɂ����ĐV�L�������ܗL���i�̌���y�ѐ��܂̏��F�\�����s���Ƃ��ɕK�v�Ȉ��萫�������т�����������
��Q1B�u�V����y�ѐV���܂̌����萫�����K�C�h���C���v��
�E�V����y�ѐV���܂����F�\������ۂɕK�v�Ƃ��������萫�Ɋւ�����邱�Ƃ�ړI�Ƃ��Ă���
��Q1C�u�V���^�o�H���i���̈��萫�������т̎戵���Ɋւ���K�C�h���C���v��
�E�V���^���i���ɂ��ď��F�\������ۂ̈��萫�������т̎戵������������
��Q1D�u����y�ѐ��܂̈��萫�����ւ̃u���P�b�e�B���O�@�y�у}�g���L�V���O�@�̓K�p�v��
�E�u�}�g���L�V���O�@�y�уu���P�e�B���O�@�v�Ɋւ���w�j����������
��Q1E�u���萫�f�[�^�̕]���Ɋւ���K�C�h���C���v��
�E���萫�f�[�^�����F�\���ɂ����Ăǂ̂悤�ɗ��p���ă��e�X�g���Ԗ��͗L�����Ԃ������悢��������������
��Q1F�u�C����III�y��IV�ɂ����鏳�F�\���̂��߂̈��萫�������тɊւ���K�C�h���C���v�̔p�~��
�E���Ăd�t���i�K�����a���ۉ�c�i�h�b�g�j�̍��ӂɊ�Â��A�h�b�g�p�P�e�K�C�h���C����p�~����
|
2003.6.3
1997.5.28
1997.5.28
2002.7.31
2003.6.3
2006.7.3 |
Q2 |
�y���͖@�o���f�[�V�����z
��Q2�iR1�j�u���͖@�o���f�[�V�����Ɋւ���e�L�X�g�i���{���ځj�v��
�E���{�C�A�����J���O���y�у��[���b�p�A���̎O�ɓ��ɂ�������i�̏��F�\���Ɋ܂܂�镪�͖@�ɂ��āC�o���f�[�V�������s���ۂɌ������K�v�ȕ��͔\�p�����[�^�ɂ��ċL�ڂ�������
��Q2�iR1�j�u���͖@�o���f�[�V�����Ɋւ���e�L�X�g�i���{���@�j�v��
�E�u���͖@�o���f�[�V�����Ɋւ���e�L�X�g�i���{���ځj�v��⊮�������
�E�X�̕��͖@�Ɋ֘A����l�X�ȕ��͔\�p�����[�^������������@�ɂ��āA���̎w�j������
|
1995.7.20
1997.10.28 |
Q3 |
�y�s�����z
�@��Q3A�iR2�j�u�V�L�������ܗL���i�̂�������̕s�����Ɋւ���K�C�h���C���v��
�E�V�L�������ܗL���i�̂����A���w�I�����@�Ő�������錴�̕s�����̗ʋy�т��̈��S���̊m�F�Ɋւ��鏳�F�\���ɍۂ��Ă̎w�j�������Ă���
�A��Q3A�iR2�j�u�V�L�������ܗL���i�̂�������̕s�����Ɋւ���K�C�h���C���̈ꕔ����v��
�E���Ăd�t���i�K�����a���ۉ�c�i�h�b�g�j�ɂ����鍇�ӂɊ�Â��A�V�L�������ܗL���i�̂�������̕s�����Ɋւ���K�C�h���C�����ꕔ����
�B��Q3B�iR2�j�u�V�L�������ܗL���i�̂������܂̕s�����Ɋւ���K�C�h���C���v��
�E�V�L�������ܗL���i�̂����A���w�I�����@�ɂ�萻������錴���p���Đ�������鐻�ܒ��̕s�����̗ʋy�т��̈��S���̊m�F�Ɋւ��鏳�F�\���ɍۂ��Ă̎w�j�������Ă���
�C��Q3B�iR2�j�u�V�L�������ܗL���i�̂������܂̕s�����Ɋւ���K�C�h���C���̉���v��
�E���Ăd�t���i�K�����a���ۉ�c�i�h�b�g�j�ɂ����鍇�ӂɊ�Â��A�V�L�������ܗL���i�̂������܂̕s�����Ɋւ���K�C�h���C�����ꕔ����
�D��Q3C�iR3�j�u���i�̎c���n�}�K�C�h���C���v��
�E���i�̐����̍ۂɂ́C��Ő��̗n�}���g�p����悤�Ɋ�������ƂƂ��ɁC�������̎c���n�}�ɂ��ēŐ��w�I�ɋ��e��������x�l�������Ă���
�E��Q3C�iR3�j�u���i�̎c���n�}�K�C�h���C�� N�|���`���s�����h���iN-Methylpyrrolidone�j��PDE�l�ɂ��ā^�e�g���q�h���t�����iTetrahydrofuran�j��PDE�l�ɂ��āv��
�E���Ăd�t���i�K�����a���ۉ�c�i�h�b�g�j�ɂ����āApermissible daily exposure�i�o�c�d�l�j�ɂ��č��ӂ��ꂽ���Ƃ���A�u���i�̎c���n�}�K�C�h���C���ɂ��āv�̈ꕔ�����߂�
�F��Q3C�iR3�j�u���i�c���n�}�̌��x�l�ɂ��āv��
�E���i�c���n�}�̌��x�l�ɂ��āA�ŐV�̉Ȋw�I�m�����Ɋ�Â������̗]�n������ꍇ�̌����̎菇������
�G��Q3C�iR5�j�u���i�̎c���n�}�K�C�h���C���v�̉�����
�E���Ăd�t���i�K�����a���ۉ�c�i�h�b�g�j�ɂ����āA�N������Permitted Daily Exposure �i�o�c�d�l�j�ɂ��č��ӂ��ꂽ���Ƃ���A�u���i�̎c���n�}�K�C�h���C���ɂ��āv�̈ꕔ�����߂�
�H��Q3C�iR6�j�u���i�̎c���n�}�K�C�h���C���v�̉�����
�E����EU���i�K�����a���ۉ�c(ICH)�ɂ����āA�g���G�`���A�~���y�у��`���C�\�u�`���P�g���� Permitted Daily Exposure (PDE �l) �ɂ��č��ӂ��ꂽ���Ƃ���A�u���i�̎c���n�}�K�C�h���C ���ɂ��āv�̈ꕔ�����߂�
�I��Q3D�u���i�̌��f�s�����K�C�h���C���ɂ��āv��
�EICH Q9�ɋL�ڂ���Ă��郊�X�N�}�l�W�����g�̌�����p���Đ��ܒ��̌��f�s������]�����A�Ǘ�����v���Z�X����������
|
2002.12.16
2006.12.4
2003.6.24
2006.7.3
1998.3.30
2002.12.25
2002.12.3
2011.2.21
2018.7.19
2015.9.30 |
Q4 |
�y��Ǖ��z
��ICH Q4B �K�C�h���C���i��Ǖ��e�L�X�g��ICH�n��ɂ����đ��ݗ��p���邽�߂̕]���y�ъ����j��
�EQ4B ���ƍ�ƕ���iEWG�j�ɂ���Ǖ��e�L�X�g�̕]���y�ъ����̃v���Z�X�������AICH �n��ɂ������Ǖ��e�L�X�g�̑��ݗ��p�𑣐i���邽�߂̂���
��Annex 1�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���M�c�������@�j��
�E���M�c�������@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 2�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���ˍ܂̍̎�e�ʎ����@�j��
�E���ˍ܂̍̎�e�ʎ����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 3�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���ˍ܂̕s�n�������q�����@�j��
�E���ˍ܂̕s�n�������q�����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 4�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���������x�����@�y�єۈ��i�̔������w�I�i�������j��
�E�ې��i�̔������w�I�����F���ې������ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 5�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i�����@�j��
�E�����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 7�iR2�j�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i�n�o�����@�j��
�E�n�o�����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 8�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���ێ����@�j��
�E���ێ����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 9�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���܂̖����x�����@�j��
�E���܂̖����x�����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 10�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������iSDS�|���A�N�����A�~�h�Q���d�C�j���@�j��
�ESDS�|���A�N�����A�~�h�Q���d�C�j���@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 11�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i�L���s�����[�d�C�j���@�j��
�E�L���s�����[�d�C�j���@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 12�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i���x����@�i�ӂ邢�����@�j�j��
�E���x����@�i�ӂ邢�����@�j�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex 13�FICH Q4B �K�C�h���C���Ɋ�Â������ʕt�������i�������x�y�у^�b�v���x����@�j��
�E�������x�y�у^�b�v���x����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex14�F�K�C�h���C���Ɋ�Â������ʕt�������i�G���h�g�L�V�������@�j��
�E�G���h�g�L�V�������@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
��Annex6: ICHQ4B�K�C�h���C���Ɋ�Â������ʕt�������i���܋ψꐫ�����@�j�ɂ��ā�
�E���܋ψꐫ�����@�ɂ��� Q4B ���ƍ�ƕ���ŕ]�����ꂽ���ʂ�����������
|
2009.5.26
2009.5.26
2010.2.8
2010.2.8
2010.9.17
2010.9.17
2011.7.26
2010.9.17
2011.1.27
2011.1.27
2011.1.27
2011.1.27
2012.11.8
2013.3.21
2014.4.17 |
Q5 |
�y������i�̕i���z
��ICH Q5A�iR1�j�u�q�g���͓����זE����p���Đ��������o�C�I�e�N�m���W�[���p���i�̃E�C���X���S���]���v��
�E�E�C���X�����̊댯����]�����A���i����E�C���X��r�����A�� ���ăq�g���͓����זE�R���̈��S�ȃo�C�I�e�N�m���W�[���p���i�����邽�߂ɂǂ̂悤�ȃA�v���[�`������悢�����������Ă���
��ICH Q5B�u�g����DNA�Z�p�����p�����^���p�N�����Y�ɗp����זE���̈�`�q�����\���̂̕��́v��
�E�^�j�זE�y�ь��j�זE�őg���� DNA �Z�p�����p���ă^���p�N���Y����ۂ̈�`�q�����\�� �̂̉�͂Ɋւ���w�j������������
��ICH Q5C�u������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�̈��萫�����v��
�E������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�̈��萫�����ɂ��āCICH �ɂ�����O�ɂ̍��ӎ����Ɋ�Â��C���̕W���I�Ǝv������@������������
��ICH Q5D�u������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�����p�זE��܂̗R���A�����y�ѓ�����́v��
�E������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�����p�זE��܂̗R���A�����y�ѓ�����͂ɂ��āAICH�ɂ�����O�ɂ̍��ӎ����Ɋ�Â��A���̕W���I�Ǝv������@������������
��ICH Q5E�u������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�̐����H���̕ύX�ɂƂ��Ȃ��������^�������]���v��
�E������i�i�o�C�I�e�N�m���W�[���p���i�^���� �N���R�����i�j�̐����H���̕ύX�ɂƂ��Ȃ��������^�������]���ɂ��āA�h�b�g �ɂ�����O�ɂ̍��ӎ����Ɋ�Â��A���̕W���I�]�����@������������
|
2000.2.22
1998.1.6
1998.1.6
2000.7.14
2005.4.26 |
Q6 |
�y�K�i����ю������@�z
��ICH Q6A�u�V���i�̋K�i�y�ю������@�̐ݒ�v��
�E�V����ƐV���܂ɂ��āA���E�K�͂ł̒P��̋K�i�y�ю������@�̐ݒ�𑣐i���邱�Ƃ�ړI�Ƃ��Ă���
��ICH Q6B�u������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j�̋K�i�y�ю������@�̐ݒ�v��
�E������i�i�o�C�I�e�N�m���W�[���p���i�^�����N���R�����i�j ��V���ɏ��F�\�����A��s��ڎw���ɓ������āA�K�i�y�ю������@�̐ݒ���тɂ��̍������\�Ȍ��荑�ۓI�ɐ������̂�����̂Ƃ��邽�߂̈�ʓI�Ȍ����ɂ��āA���炩�ɂ������̂ł���
|
2001.5.1
2001.5.1 |
Q7 |
�y����GMP�̃K�C�h���C���z
�EICH Q7�i���ӓ�����Q7A�j�ł̍��ӂɊ�Â��A����i���i�̗L�������j�Ɋւ��鐻���Ǘ��y�ѕi���Ǘ��i�f�l�o�K���j�̎��{�ɂ��āA���̕W���I�Ȃ����������������
�EICH Q7�̉��߂ɂ�萶����s�m�����m�������AQ&A�W���A2016�N�ɒʒm���ꂽ
|
2001.11.2
2016.3.8 |
Q8 (R2) |
�y���܊J���i����j�z
�E���i����ѐ����H���̊J���ɑ���A�Ȋw�ƃ��X�N�Ɋ�Â��A�v���[�`�ɂ��ċL�q
�E�f�U�C���X�y�[�X�A�K���̒e�͓I�Ȏ��g�݂Ƃ����T�O��
�EQuality by Design�iQbD�j�̊T�O�����AQbD�J���A�v���[�`�ƃf�U�C���X�y�[�X�̗���
�E�N�I���e�B�E�o�C�E�f�U�C���iQbD) �́A���O�̖ڕW�ݒ�Ɏn�܂�A���i�y�эH���̗�����тɍH���Ǘ��ɏd�_���������A�����ꂽ�Ȋw�y�ѕi�����X�N�}�l�W�����g�Ɋ�Â��̌n�I�ȊJ����@
|
2010.6.28 |
Q9 |
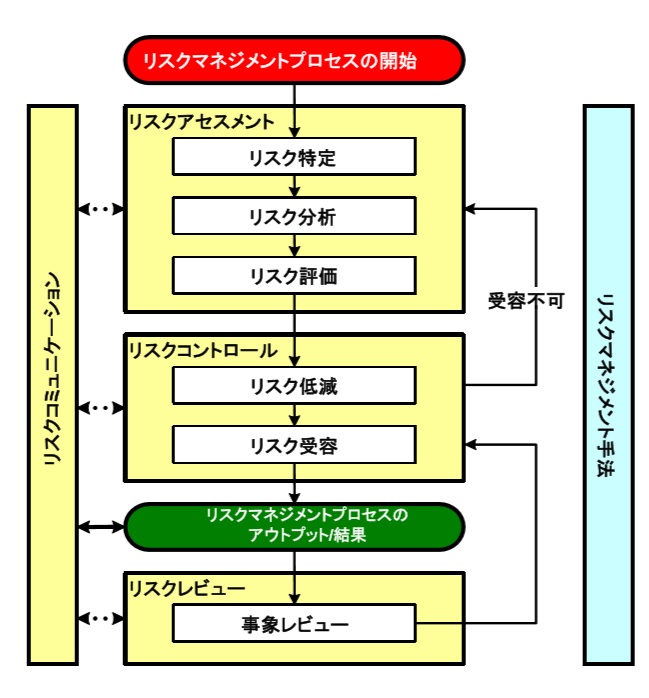
�y�i�����X�N�}�l�W�����g�z
�E�i���ɑ��郊�X�N�̃A�Z�X�����g�A�R���g���[���A�R�~���j�P�[�V��������у��r���[�ɑ���n���I�ȃv���Z�X�ɂ��ċL�q
�E���i���C�t�T�C�N���i�J���A�����A���ʁj��ʂ��ēK�p�����
�E�i�����X�N�}�l�W�����g�Ɋւ��錴���A���@�_����уc�[ ���̗���܂�
�E�i���ɑ��郊�X�N�̃A�Z�X�����g�́A�u�Ȋw�I�m���Ɋ�Â��Ă��邱�Ɓv�A�u���҂̕ی�Ɍ��т��Ă��邱�Ɓv�A�u���i�̃��C�t�T�C�N���S�̂ɂ���Ԃ��Ɓv���K�v
�E�i�����X�N�}�l�W�����g�̑̌n�I�ȃA�v���[�`����邱��
�@
|
2006.9.1 |
Q10 |
�y���i�i���V�X�e���z
�E�K�ȕi��������L���鐻�i���������邽�߂̃V�X�e�����m�����A���{���A�ێ����邱��
�E�����v���Z�X�̔\�͂̕ۏ���邱��
�E�p���I���P�𑣐i���邱��
�EPQS�̗v�f�ɂ́A�u���j�^�����O�V�X�e���v�A�u�����[�u�y�ї\�h�[�u�iCAPA�j�V�X�e�� �v�A�u�ύX�}�l�W�����g�V�X�e�� �v�A�u�����v���Z�X�̉ғ����\�y�ѐ��i�i���̃}�l�W�����g���r���[�v���܂܂�� |
2010.2.19 |
Q11 |
�y����̊J���Ɛ����i���w�����^�����N���R�������j�z
�EICH Q8�AQ9�AQ10�iQ�g���I�j�ɋL�q����Ă��錴���ƊT�O���A����̊J���Ɛ����Ɋ֘A���Đ�����������
�E���ޗ��Ǘ����猴��A���܂Ɏ���܂ł̈�т����Ǘ��헪�\�z�̂���Ȃ鑣�i
�E�o�������y�ѐ����N�����ޗ��I��ɌW�鋤�ʗ����̑��i
|
2014.7.10 |
�@
| �����̒m�� | GMP�E�i���Ǘ��̒m�� |
|---|---|
|
���i�����̗��ꓙ |
GMP�Ƃ�!?�� |


| �N���[���E���R�[���̒m�� | ���i�̒m�� |
|---|---|
|
�N���[���E������ᓙ |
�ǂ����i�Ƃ́I�H�� |


| ���ޗ��E����E������̒m�� | �ݔ��E���̒m�� |
|---|---|
|
����Ƃ́I�H�� |
�����ɓK������Ǝ��Ƃ́I�H�� |


| �V���g�s�b�N�X | ����ƍ�ƈ��̒m�� |
|---|---|
|
�V���̘b��E���� |
��ƈ��ɋ��߂��邱�ƂƂ́I�H�� |


�֘A���|�[�g
- GMP�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP�Ƃ́I�H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- GMP�̂R�����Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP�R�̊�{�v���Ƃ́HGMP�R�����Ƃ́I�H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- GMP�ȗ߁i�����j
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP�ȗ߁i�����j�Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- GMP�ȗ߂̎戵���i�����j
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP�ȗ߂̎戵���i�����j�Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- �Ȃ�GMP���K�v�Ȃ̂��I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�Ȃ�GMP���K�v�Ȃ̂��I�H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- �]�ƈ���GMP����邽�߂ɂ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B��Ǝ҂�GMP����邽�߂ɂ́H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- GMP���@�̎w�E����́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP���@�̎w�E����́I�H�����|�[�g�ɂ܂Ƃ߂܂����B
- GMP���ᔽ������ǂ��Ȃ�̂��I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGMP���ᔽ������ǂ��Ȃ�̂��I�H���i������H���ő�Ȃ��ƃ��|�[�g�ɂ܂Ƃ߂܂����B
- �K�v�Ȏ菇����
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�K�v�Ȏ菇�����́H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- GQP�ȗ߁i�����j
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BGQP�ȗ߂Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- �f�[�^�C���e�O���e�B�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�f�[�^�C���e�O���e�B�Ƃ́H���i�������ő�Ȃ��Ƃ����|�[�g�ɂ܂Ƃ߂܂����B
- �o���f�[�V�����Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�o���f�[�V�����Ƃ́I�H���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- �N�I���t�B�P�[�V�����Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�N�I���t�B�P�[�V�����Ƃ́I�H���i������H���ő�Ȃ��Ƃ����|�[�g�ɂ܂Ƃ߂܂����B
- �o���f�[�V�����̋L�^
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B�M�p�ł���L�^�Ƃ́I�H���i������H���ő�Ȃ��ƃ��|�[�g�ɂ܂Ƃ߂܂����B
- ICH�K�C�h���C���Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���i�K���K�C�h���C���uICH�v�Ƃ́I�H�����|�[�g�ɂ܂Ƃ߂܂����B
- ���i�i���V�X�e���uICH Q10�v�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���i�i���V�X�e���uICH Q10�v�Ƃ́I�H��Ȃ��Ƃ����|�[�g�ɂ܂Ƃ߂܂����B
- PIC/S�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BPIC/S�Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- PMDA�ƌ����J���Ȃ̈Ⴂ�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��BPMDA�ƌ����J���Ȃ̈Ⴂ�Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
- ���ʍ����@�Ƃ́I�H���ʍ����@�ł̎w�E����
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���ʍ����@�Ƃ́I�H���ʍ����@�ł̎w�E��������|�[�g�ɂ܂Ƃ߂܂����B
- ��E�i�����j�Ƃ́I�H
- ���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B��E�i�����j�Ƃ́I�H���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B