
���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���{�̈��i�̖@�̌n�ɂ��Ă܂Ƃ߂܂����B
��@�@�Ƃ́I�H��@�@�����ɂ���
��@�@�Ƃ́I�H
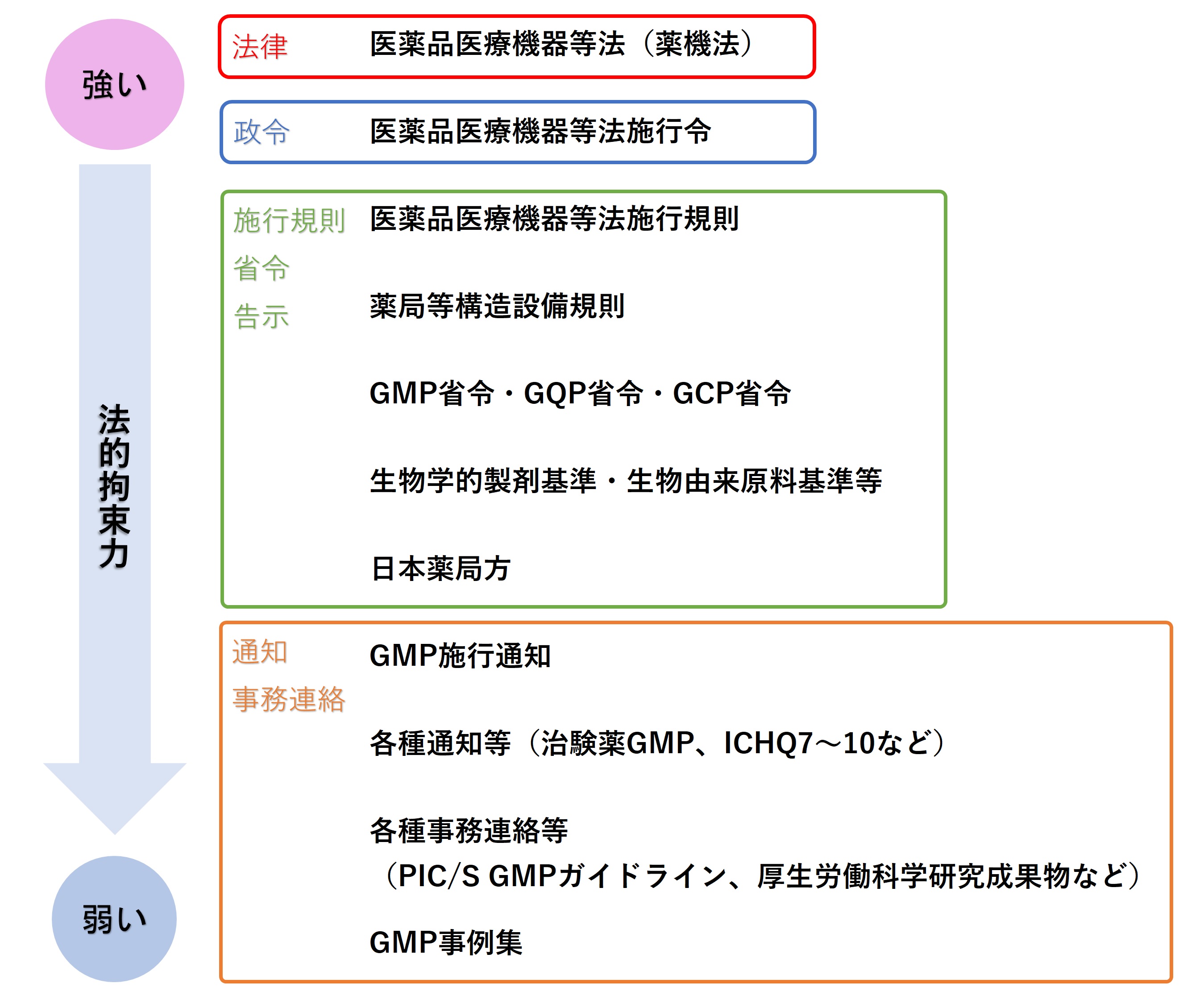
�@��@�@�Ƃ́A�u���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���v�̂��ƂŁA���i��Ë@�퓙�@�A��@�@�Ɨ������B
�@���{�ɂ�������i�A��O�i�A���ϕi�A��Ë@��y�эĐ���Ó����i�Ɋւ���^�p�Ȃǂ��߂��@���ł���B
�@���蓖���́u�@�v�i�₭���ق��j�ł��������A2014�N�ɖ@���̈ꕔ����������@���̎{�s�ɂ�茻�݂��u��@�@�v(������ق�)�ɉ��߂�ꂽ�B
�@��@�@�ł́A�s���̏��F��m�F�A���A�ē��̂��ƂłȂ���A���i���O�i�A���ϕi�A��Ë@��̐�����A���A���܂ʼnc�Ƃ��Ă͂Ȃ�Ȃ��悤��߂Ă���B
��@�@�����ɂ���
�@���i��Ë@�퓙�@�i��@�@�j�̉����́A��ËZ�p�̐i����Љ�I�ۑ�ɑΉ����邽�߂ɍs���Ă���B
�@�@�����ɂ���Ĉ��i���Ë@��A�Đ���Ó����i�̈��S����L�����̊m�ہA���҂ւ̑����Ȃǂ�ڎw���Ă���B
�@2019�N12��4���ɉ�����@�@�����z����A2020�N9��1�����A������@�@�̊֘A�ȗ߂��{�s���ꂽ�B�@
�@������@�@�̓��e�́A4�̑區�����琬��B
�@
�@���i�A��Ë@�퓙�������S�E�v���E�����I�ɒ��邽�߂��J������s�̌�܂ł̐��x���P
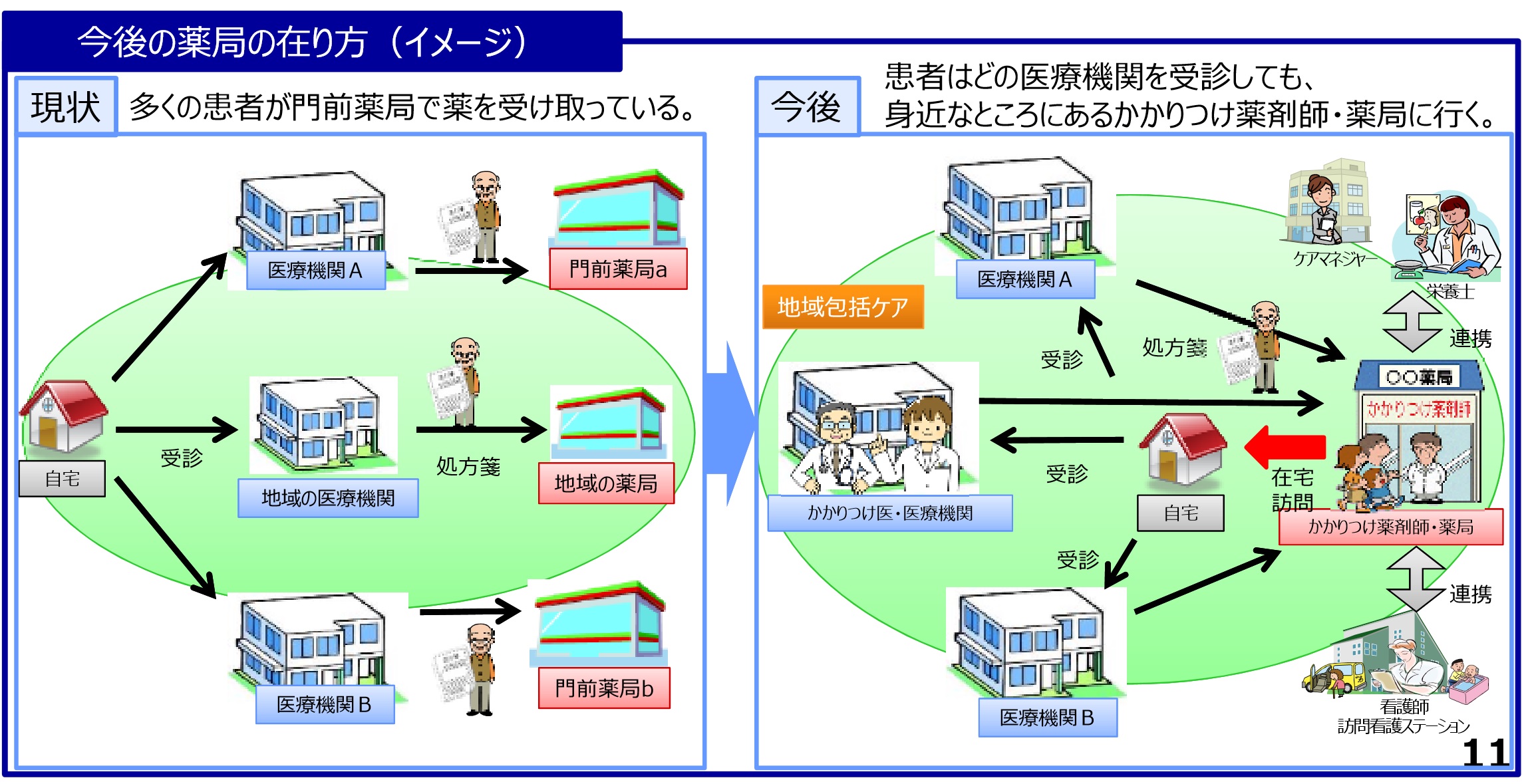
�A�Z�݊��ꂽ�n��Ŋ��҂����S���Ĉ��i���g�����Ƃ��ł���悤�ɂ��邽�߂���t�E��ǂ̂�����̌�����
�B�M���m�ۂ̂��߂��@�ߏ���̐����̐���
�C���̑�

��@�@�������e�ɂ���
�@�@���i�A��Ë@�퓙�̊J������s�̌�܂ł̐��x���P
�@�E�����J���Ȃ̒ʒm�Ɋ�Â��ĉ^�p����Ă���u��삯�R���w�萧�x�v�A�u�����t���������F���x�v��@���Ɋ�Â����x�Ƃ���B
�@�E�u��삯�R���w�萧�x�v�́A���E�ɐ�삯�ē��{�Ő\�����悤�Ƃ������I�V������F�R���ŗD�����鐧�x�B���i��Ë@�푍���@�\�iPMDA�j���\�����������O�ɕ]�����邱�ƂŎ����I�ɐR����O�|�����A�ʏ�12�J��������R������6�J���ɒZ�k����B
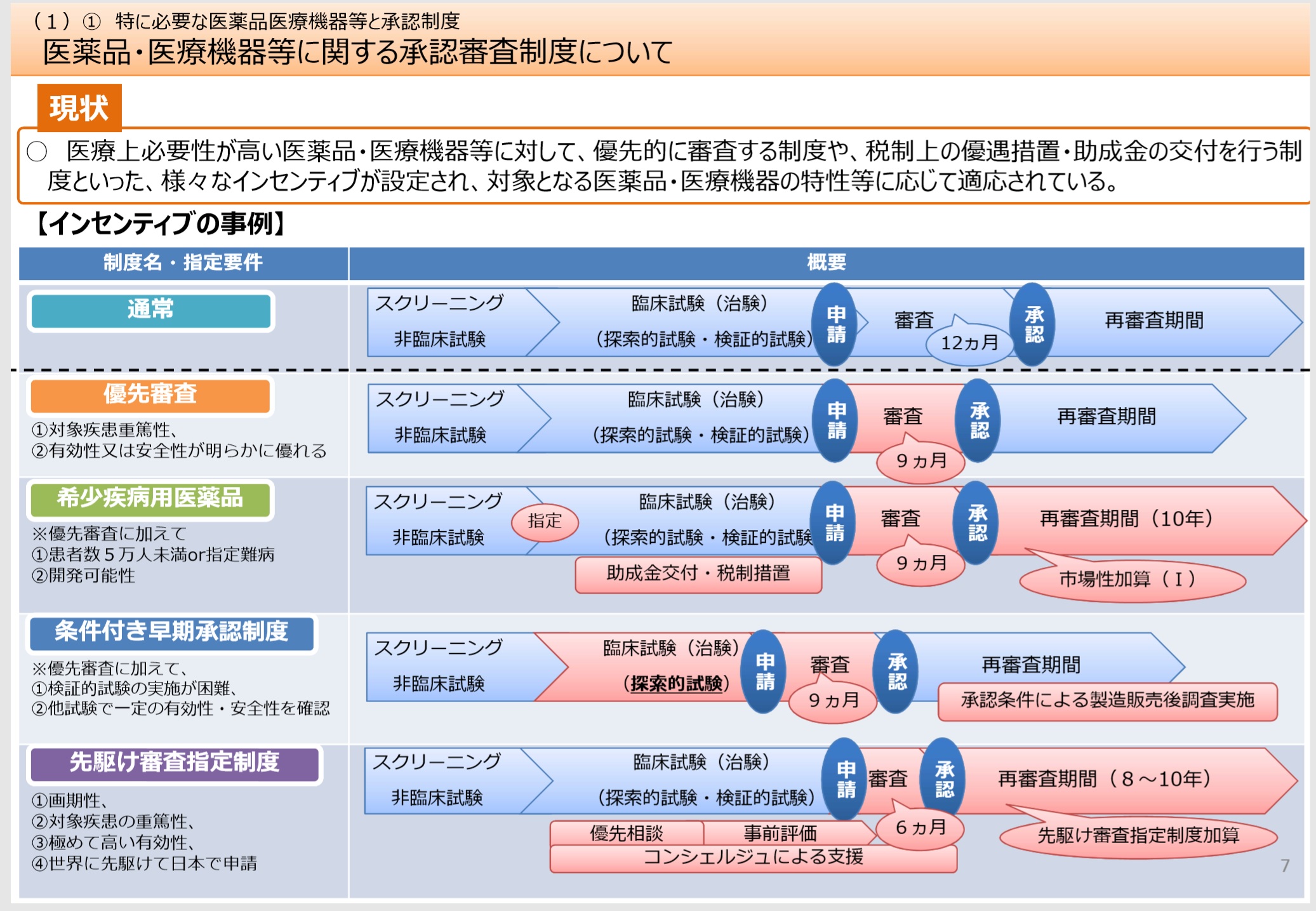
�@�E�u�����t���������F���x�v�́A�d�ĂŗL���Ȏ��Ö@���Ȃ������A���Ґ������Ȃ��Ȃǂ̗��R�Ō��ؓI�Տ������i��ʓI�ɂ͗Տ���3���qP3�r�����j���s���̂�����������ΏۂƂȂ�B
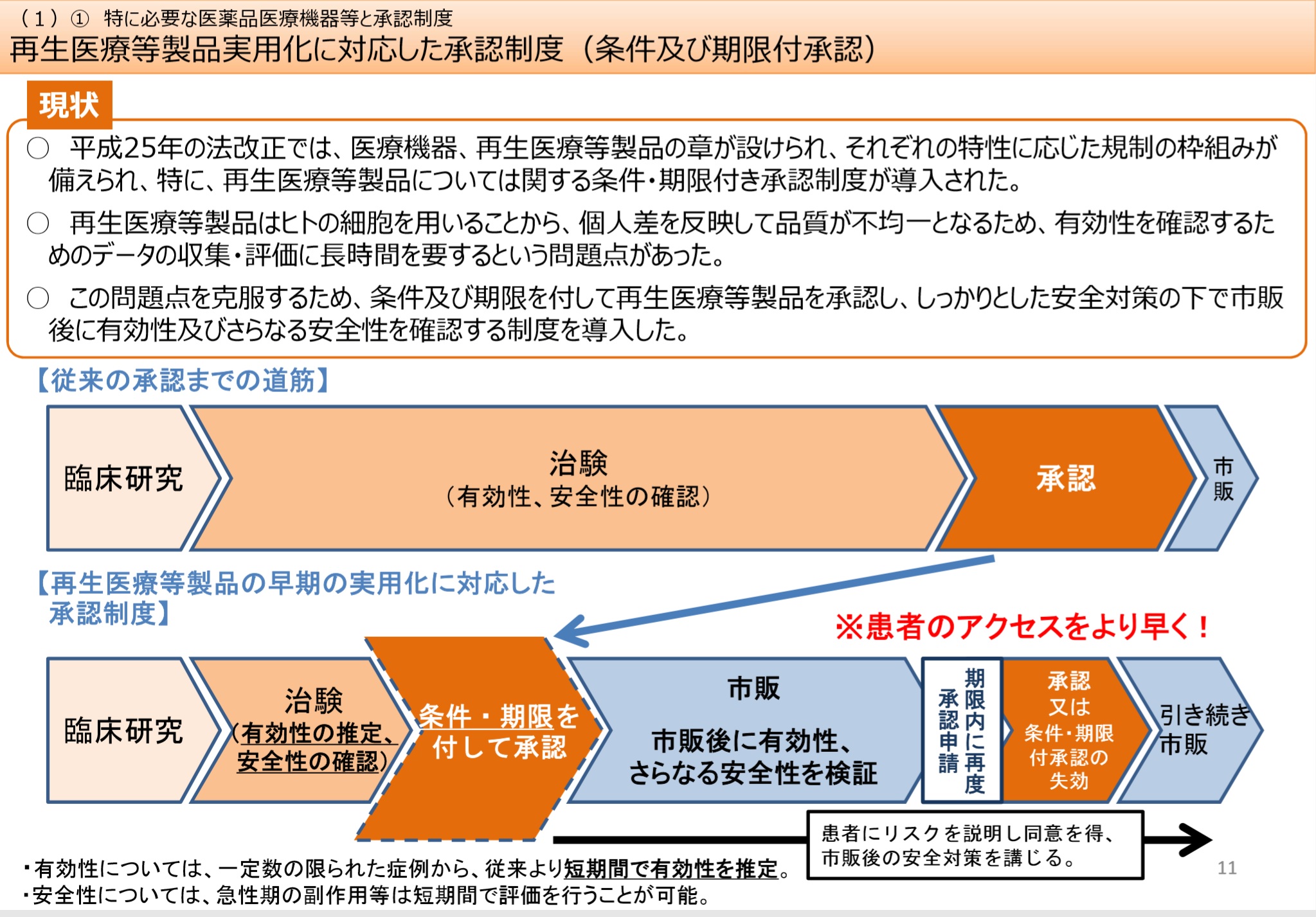
�@���ؓI�����ȊO�̎����ŗL�����E���S����������x�������A������ɗL�����E���S�������炽�߂Ċm�F���邱�Ƃ������ɁA���ؓI�������s��Ȃ��Ă����F�������x�ł���B
�@�E�ŏI�I�Ȑ��i�̗L�����A���S���ɉe�����y�ڂ��Ȃ����i���̐������@���̕ύX�ɂ��āA���O�Ɍ����J����b���m�F�����v��ɉ����ĕύX����ꍇ�ɁA���F������͏o���Ɍ������B
�@�E�g���[�T�r���e�B����̂��߁A���i ���̕���ւ�QR�R�[�h��o�[�R�[�h���̕\���̋`���t���ȂǁA���i�A��Ë@�퓙�������S�E�v���E�����I�ɒ��邽�߂̊J������s�̌�܂ł̐��x���P���s���B
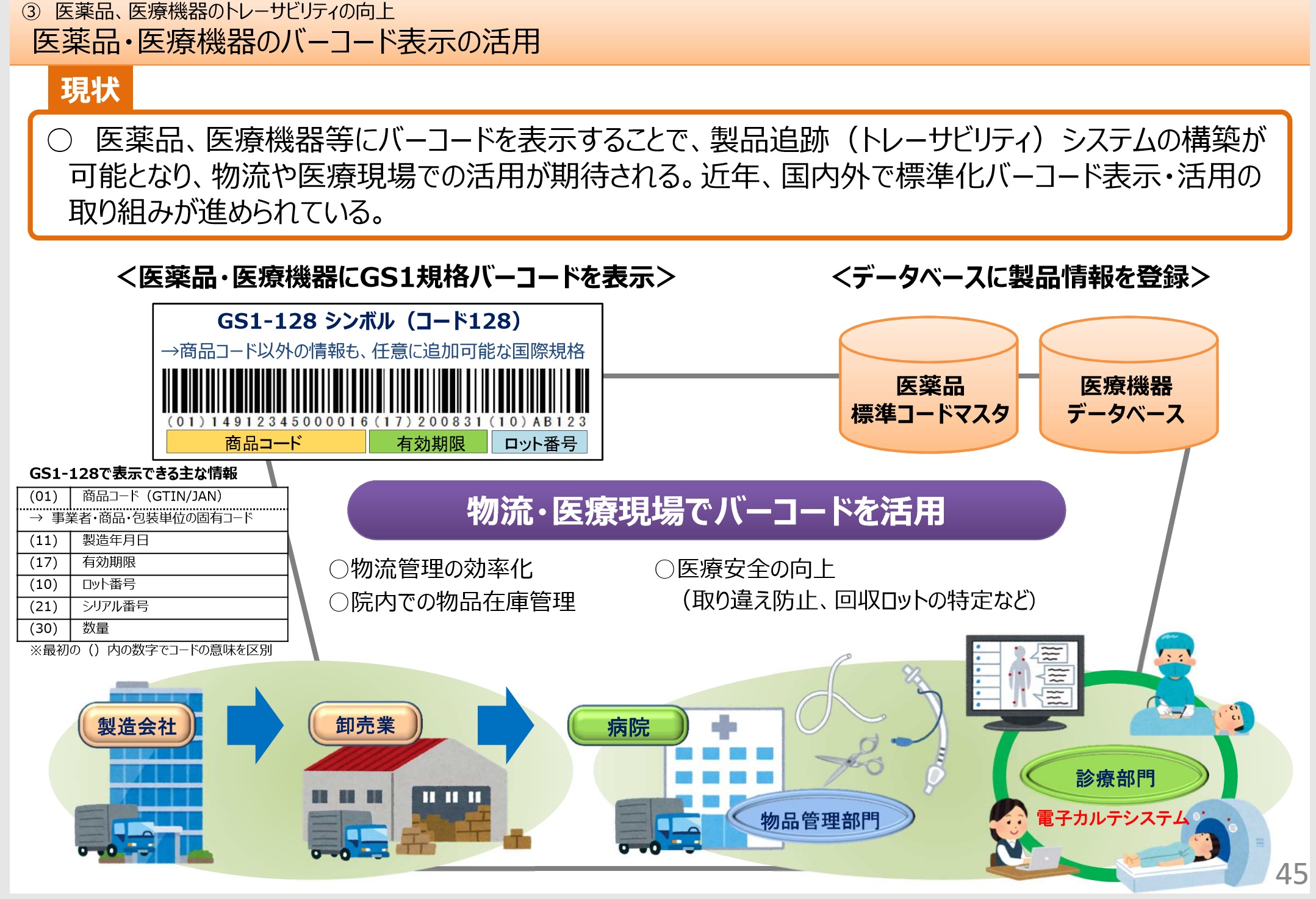

�@�E���i�[�i���̎��œ�������Ă����×p���i���Y�t�����̓�����p�~����B��Ì���ɑ���ŐV���̒͌����d�q�����邱�ƂƂȂ�B
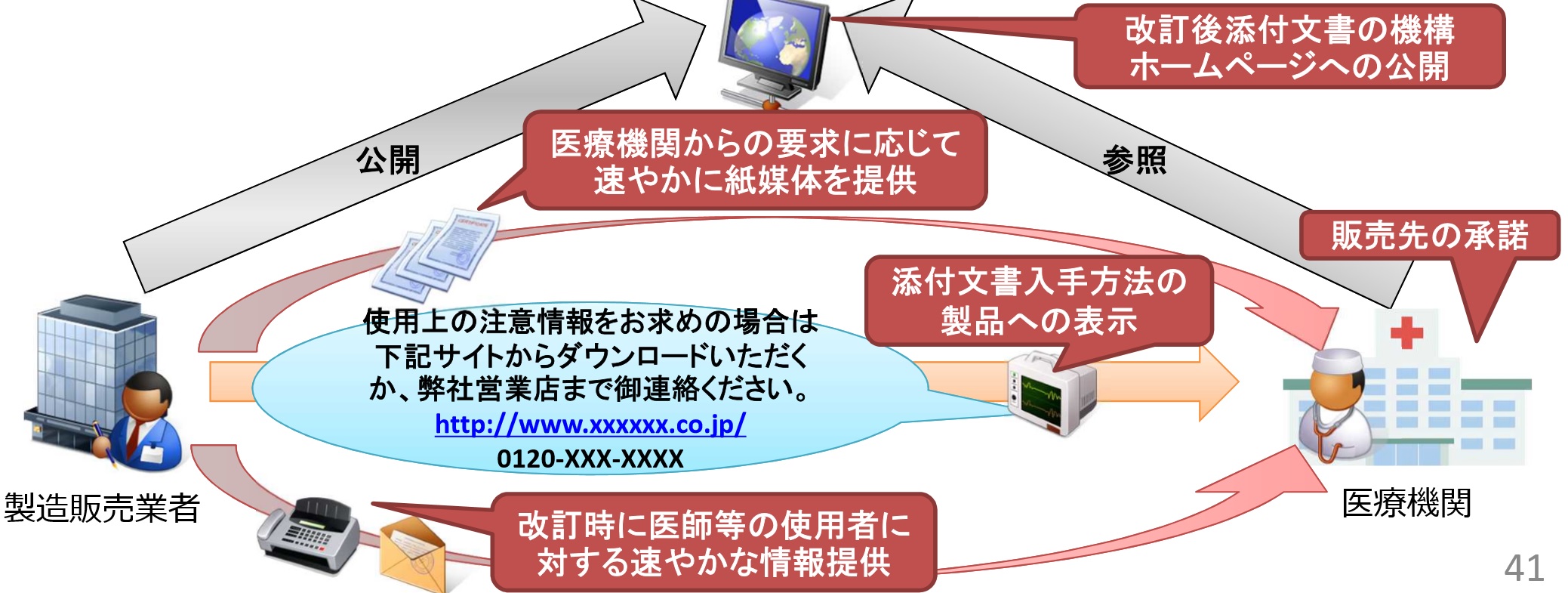
�@����ɔ����A�X�}�[�g�t�H����^�u���b�g�Ȃǒ[�������p���A��Ë@�ւ��Ǔ��ɏ����m���ɓ͂���d�g�݂̍\�z�����߂���B
�@�Y�t�����̉����͕p�ɂɍs���Ă���A��ɍŐV�̂��̂ɃA�N�Z�X�ł���悤�ɂ���̂��_���ł���B
�@�E���ۓI�ȋK���̐��������m�ۂ��邽�߁AGMP�K����������QMS�K�����������������B
�@�A��t�E��ǂ̂�����̌�����
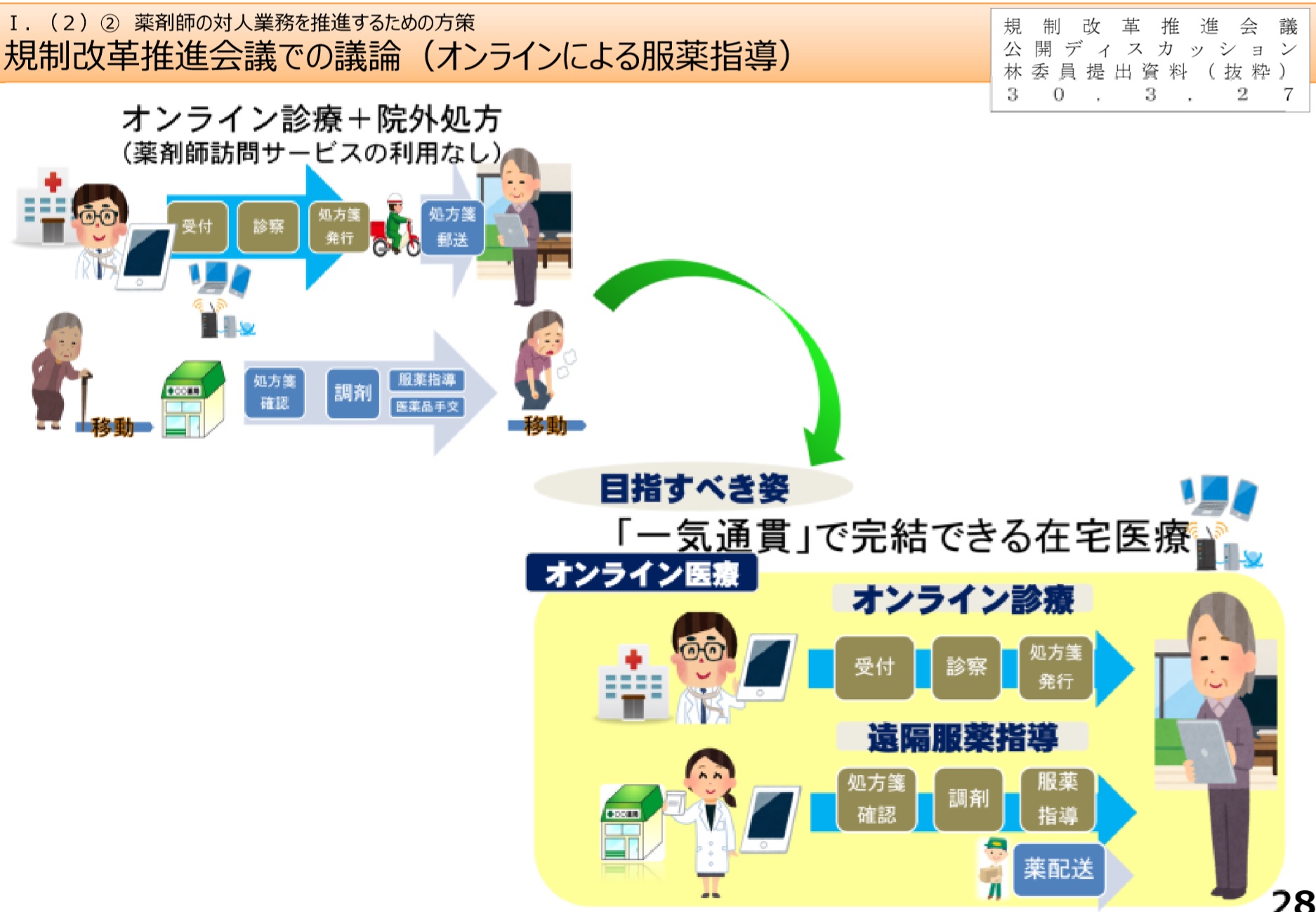
�@�E��t���A�����Ɍ��炸�A�K�v�ɉ����Ċ��҂̖�܂̎g�p�̔c��������w�����s���`����@��������B
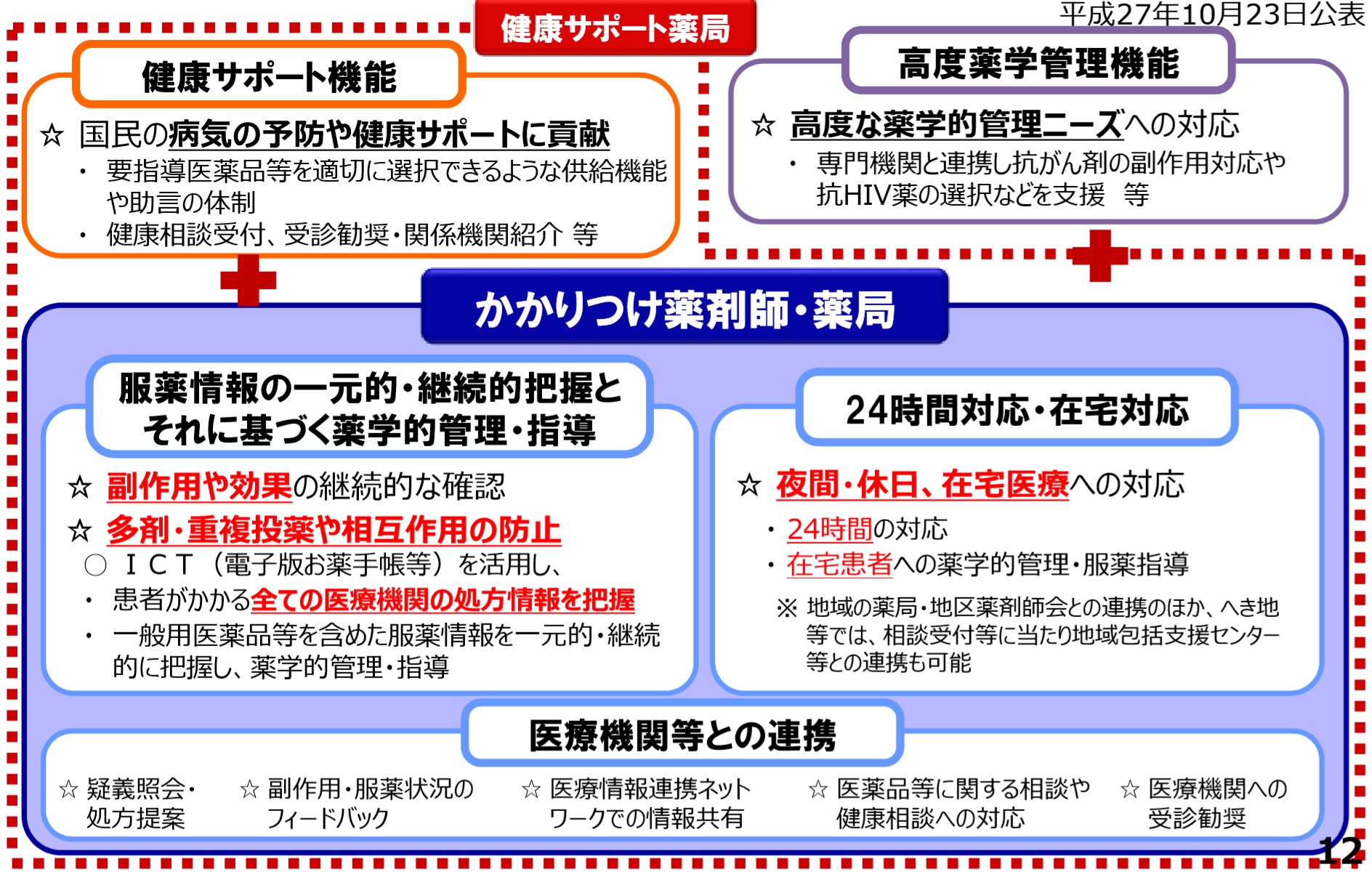

�@�E���҂����g�ɓK������ǂ�I���ł���悤�A�@�\�ʂ̖�ǂ̓s���{���m���̔F�萧�x�i���̓Ɛ�j������B
�@�E����w���ɂ��āA�Ζʋ`���̗�O�Ƃ��āA���̃��[���̉��ŁA�e���r�d�b�ɂ�镞��w�����K�肷��B
�@�B�@�ߏ���̐����̐���
�@�E�W���Ǝ҂̏d��Ȗ@�߈ᔽ�œ��ɉ��P���K�v�ȏꍇ�̖����ύX���߂�A���U�E�֑�L���ɂ����i���̔̔��ɌW��ے������x�̑n�݂ȂǁA�W���Ǝ҂ɂ��M���m�ۂ̂��߂̖@�ߏ���̐����̐������s���B
�@�E�V���ɐ݂���ے������x�ł́A���U�E�֑�L�����s���������ƂȂǂɑ��A�ᔽ�s�ׂ��s�������Ԃ�ΏۂɁA�Y�����鐻�i�̔��㍂��4.5%���ے����Ƃ��Ē�������B

�@����I�Ɉᔽ��\�������ꍇ�͉ے������Ɍ��z����ق��A�Ɩ����P���߂�Ɩ���~���߂��s�����ꍇ�͉ے����̔[�t�𖽂��Ȃ����Ƃ�����B
�@�C���̑�
�@�E���i���s���]���E�Ď��ψ���̐ݒu�B
�@�E���t�@���������A�Ȋw�Z�p�̔��W�܂����̌����̐����̊ɘa���s���Ȃǂ̌��������s���B
�@
�����J���Ȃ̉�����@�@������������
�@
�����J���ȁu��@�@�S���v��������