医薬品製造に関わる基礎知識をまとめて紹介します。医薬品・医療機器における「クレーム(品質苦情)」の定義と実務での対応プロセスを解説。
不正製造の事例(睡眠剤混入)
K製薬会社の睡眠剤混入回収事例とは!?
K製薬会社は、2020年(令和2年)12月に、爪水虫など皮膚病の治療に使う経口抗真菌剤を約10万錠分を自主回収(クラスI)すると発表した。
爪水虫などの治療に使われるジェネリック医薬品「イトラコナゾール錠50MEEK」で、最大投与量の2.5倍にあたる睡眠導入剤「リルマザホン」が誤って混入した。
また、主成分の含有量が違う同錠の「100」と「200」についても、国が承認した手順で製造されていなかったことが判明し、自主回収を行った。
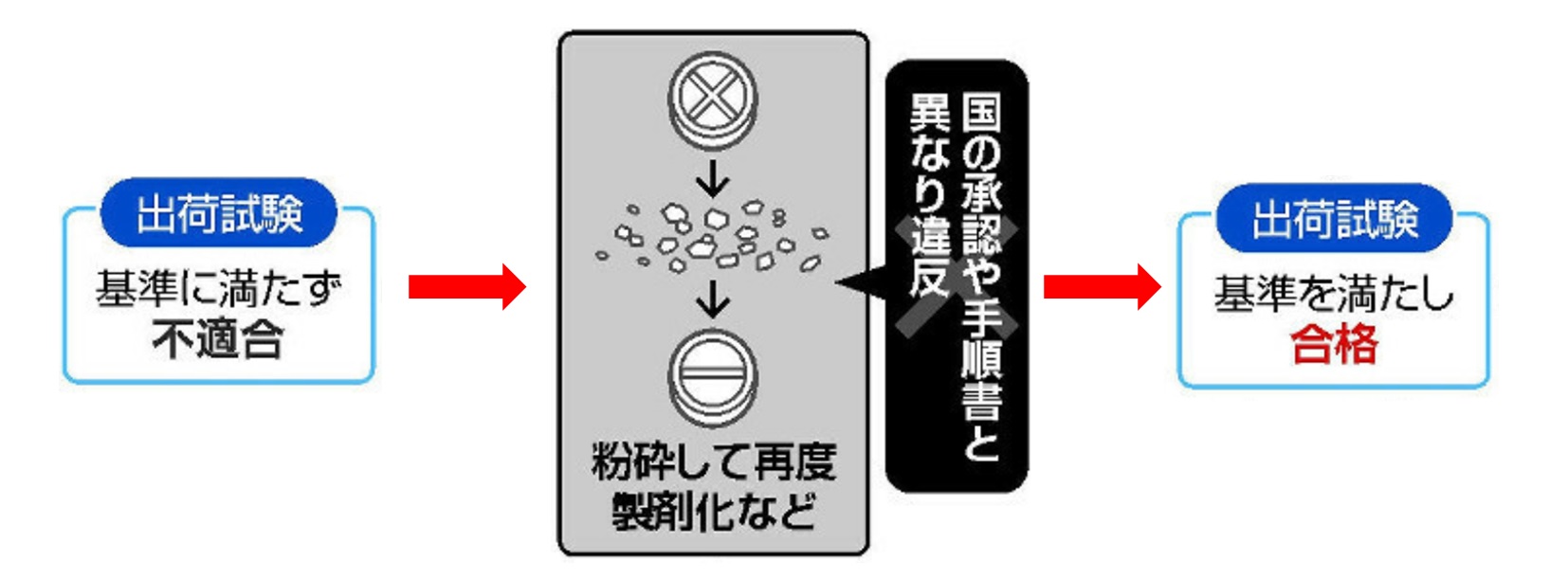
その後、厚生労働省などの立ち入り調査で出荷前の検査が不十分だったことが判明したため、自主回収は16製品に拡大した。
そして、同社による再調査の結果、規格に適合していなかったり、国の承認を得ていない方法で製造したりしていたことなどが判明したため、22製品を新たに自主回収すると発表。
同社の自主回収は計38製品に拡大した。
健康被害
睡眠剤が混入した錠剤は、全国31都道府県の364人に処方されており、服用した245人が健康被害を訴え、少なくとも1人が死亡した。
混入により、錠剤を服用した患者に意識消失や記憶喪失、ふらつきなどの健康被害が相次いだ。
混入経緯
混入は2020年7月ごろ、担当者が製造過程で目減りした原料を補充しようと、継ぎ足した際に起きた。
K製薬会社はこのとき、本来入れるべき主成分が入った容器と、睡眠導入剤成分が入った別の容器を取り違えたと説明する。
だが、本来の成分が入っていたのは、ドラム缶のような形をした大きな厚紙の容器で、間違えたのは小さな四角形の金属製と形状が大きく異なる。
K製薬会社の社長は「一般的な感覚からすれば、取り間違えることがない」とずさんさを認めざるを得なかった。
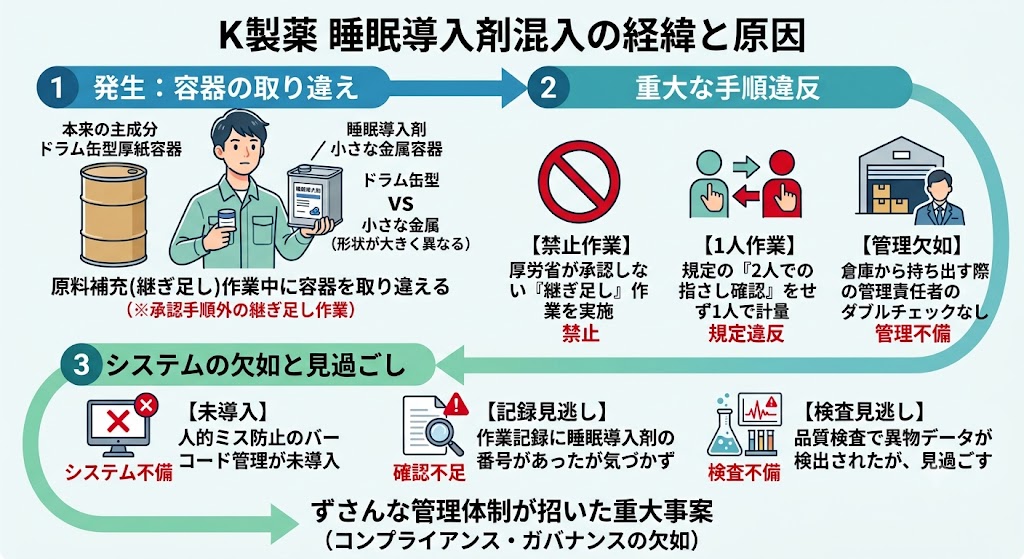
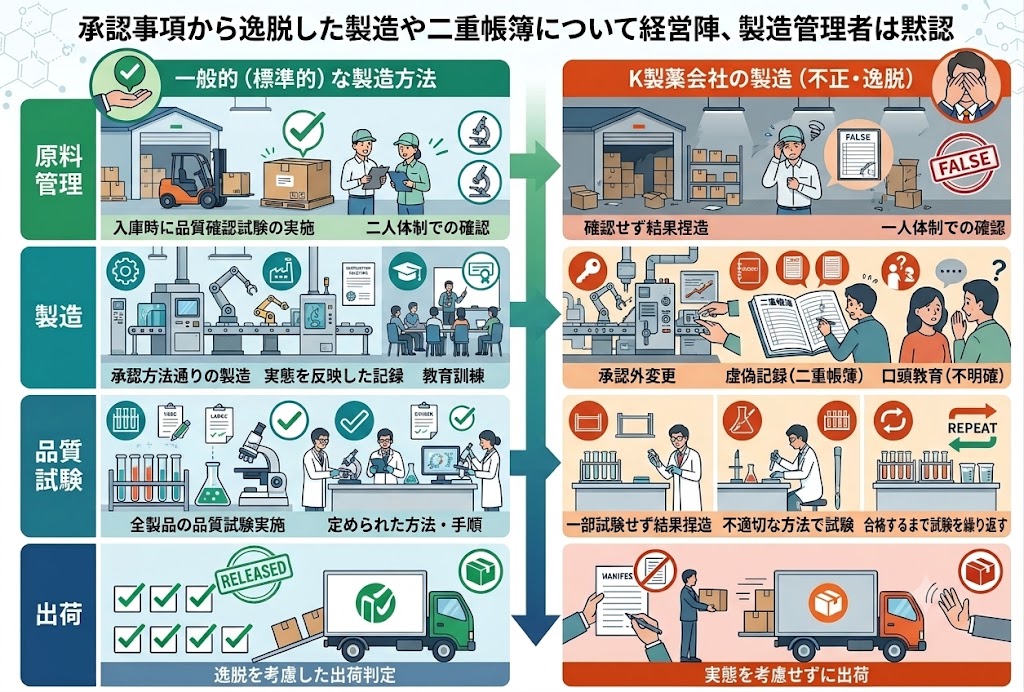
機械への付着などで、原料が減ることはあるものの、そもそも継ぎ足し作業は厚生労働省が承認した製造手順では認められていなかった。
通常、原料は倉庫内で保管し、工程に必要な分だけ管理責任者がダブルチェックした上で持ち出す。
同社の規定でも、原料を取り出す際や計量する際は2人での指さし確認が必要となっていたが、当時は担当者1人で作業していた。
当時作業にあたったのは、入社数年の若手男性従業員とみられる。従業員は「記憶がはっきりしない」との趣旨の説明をしている。
人的ミスを防止するため、バーコードを使ったコンピューター管理が業界では一般的になっているが、同社では別の工場ではバーコードを取り入れていたが、この工場では導入されていなかった。
製薬業界の関係者は「製造品目が少ない会社ならともかく、多品種の薬を製造するK製薬会社で、コンピューター管理されていないとは思わなかった」と驚く。
ミスに気付くことができた機会はほかにもあった。
製品の作業記録には本来なら投入されるはずがない睡眠導入剤成分を示す番号が記載されていたほか、最終的な品質検査でも異物混入を示すデータが検出されていたが、見過ごされた。
K社はジェネリック(後発薬)製造の中堅メーカーで、先発薬よりも飲みやすくするなど改良を加えた「付加価値製剤」の開発に注力してきた。
今回の睡眠導入剤が混入した経緯において、製造過程でのミスや手順違反が次々と明らかになった。
厚労省幹部は「この件は、コンプライアンス(法令順守)やガバナンスの問題。もし会社側が従業員のヒューマンエラーと認識しているならば、そのこと自体がゆゆしき事態だ」と批判した。
「裏手順書」が存在した
問題の薬の製造では、「裏手順書」が十数年前から製造現場で使われていた。
国が承認した手順書では、薬の主成分を全て1回で入れることになっているが、「裏手順書」では2度に分けて入れると記載されていた。
錠剤を固まりやすくするためとみられ、製造現場で十数年前から採用されていたという。
問題の薬では、従業員が主成分を2度目に入れようとして、睡眠導入剤成分と取り違えていた。
県は同社に対し、年に2〜4回程度の立ち入り調査を行ってきたが、その際は国が承認した正規の手順書のみが示されていた。
経営陣や製造管理者はこうした製造現場の実態を把握しており、二重帳簿は遅くとも平成17年には認識したが、いずれも黙認していたという。
社長は、承認外の手順で製造したことや、一部の品質検査をしなかったことについては「ルールを守ることより、作業効率を優先した」などと釈明を繰り返した。
過去最長116日間の業務停止命令
県が医薬品医療機器法に基づき、製薬会社としては過去最長となる116日間の業務停止命令を出した。
県は、違法な製造手順が常態化し、健康被害につながった事態を重く見て、行政処分で最も重い「許可取り消し」に次ぐ業務停止が妥当だと判断。期間も、県の処分基準で上限となる116日間とした。
K製薬会社は、事実上の廃業へ
2021年(令和3年)12月に、ジェネリック医薬品(後発薬)大手の会社が、K製薬会社の全工場や従業員を譲り受けると発表した。
K製薬会社は、事実上の廃業となった。
商品在庫や特許などはK製薬会社に残り、今後も健康被害を受けた人たちへの補償などを続ける。
問題から約1年、ずさんな製造管理の代償は資産譲渡にまで及んだ。
【ポイント】
1. 製造販売承認書からの逸脱と変更管理プロセスの無視
現場で「裏手順書」が十数年間も使用され、国の承認と異なる「2回払い出し」が行われていたことは、GMP(製造管理及び品質管理の基準)の根本的な逸脱である。錠剤の成形性を高める目的であっても、製造手順の変更には「変更管理手続き」を経て、検証(バリデーション)を実施し、承認書の変更申請を行う法的義務がある。現場の独断による手順改ざんを防ぐため、定期的な現場監査と、製造販売承認書と実際のSOP(標準作業手順書)の整合性を日常的に検証する仕組みの構築が必要である。
2. ヒューマンエラーを防止するハード対策(ポカヨケ)の欠如
形状や材質が全く異なる原料容器を取り違え、さらに社内規定である2名でのダブルチェックを怠り1名で作業したことが直接の引き金となった。個人の注意義務(ソフト対策)だけに頼る製造管理は限界があり、重要工程にはバーコードリーダーを用いた「原料照合システム」など、物理的に取り違いを防ぐ「ポカヨケ(Poka-Yoke)」の導入(ハード対策)が必須である。また、チェックを電子システム(MESなど)で強制し、2名の承認がなければ次の工程に進めないシステム構成が求められる。
【用語解説】
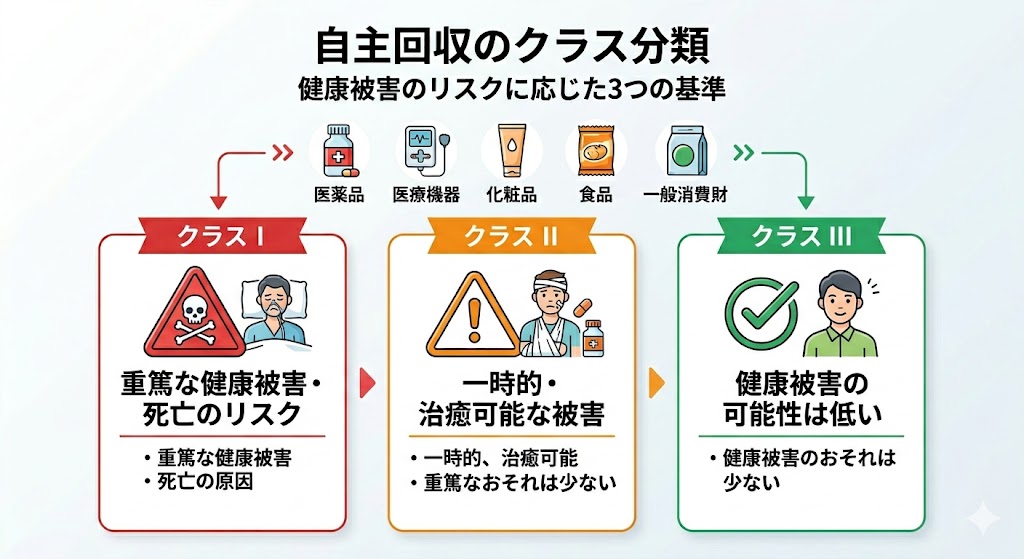
| 自主回収(クラスI) | 危篤な健康被害または死亡を引き起こす可能性がある医薬品などを回収する、最も緊急度と重要度が高い行政上の区分。 |
|---|---|
| 製造販売承認書 | 医薬品の名称、成分、分量、製造方法、効能・効果などを国(厚生労働省)が審査し、製造販売を承認した内容を記した公的文書。これに記載された手順からの逸脱は違法行為となる。 |
| 裏手順書 | 国の承認を得た「正規の手順書」とは異なり、製造現場が効率向上や作業の都合を優先して独自に作成・運用していた非公式の作業マニュアル。 |
厚生労働省の本事例に関する「プレスリリース」はこちら