���i�����Ɋւ���b�m�����܂Ƃ߂ďЉ�܂��B���ޗ��̔����Ǝ���ꌟ���ɂ��Ẵ��|�[�g�B���i�̕i���Ǘ��A���i������H���ő�Ȃ��Ƃ��܂Ƃ߂܂����B
�i���Ɋւ���K�C�h���C���uICH Q�V���[�Y�iQ1�`Q14�j�v�Ƃ́I�H
���i�̃O���[�o���ȊJ���ƈ��苟�����i�ތ���ɂ����āA�i���m�ۂ̍��ۓI�ȕW�����͕s���ł���BICH�i���i�K�����a���ۉ�c�j����߂�K�C�h���C���̂����A�uQ�iQuality�F�i���j�v�Ɋւ���V���[�Y�́A���i��������ѕi���Ǘ��̍������Ȃ��d�v�ȋK�͂ł���B
�i���Ɋւ���K�C�h���C���́AICH Q1����n�܂�A2026�N5�����݂ł�ICH Q1�`Q14�܂ō��肳��Ă���B
ICH��Q�V���[�Y�̈ʒu�Â�
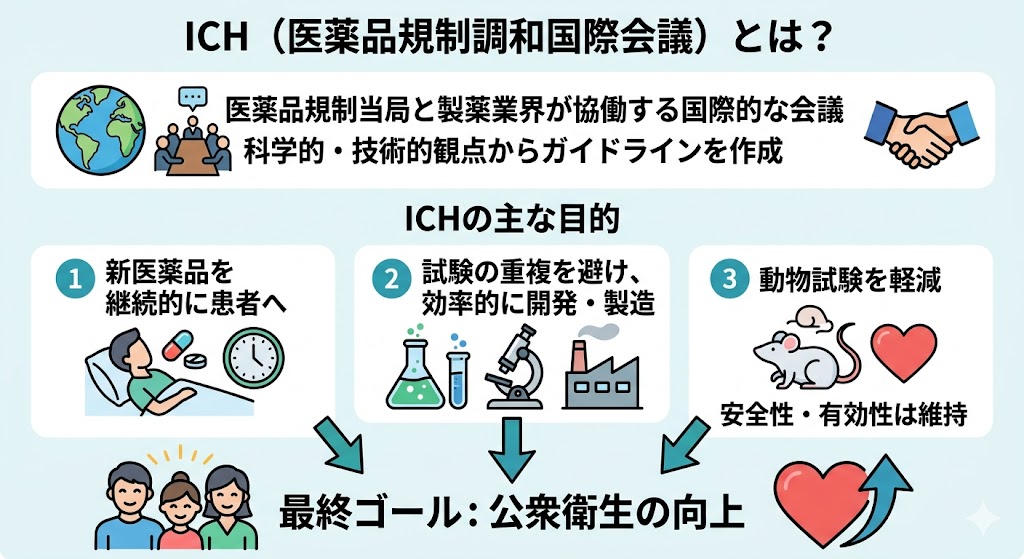
ICH�Ƃ́A���{�A�č��A���B�̋K�����ǂƐ���ƊE���������A�V��̏��F�R��������ۓI�ɒ��a���邽�߂̑g�D�ł���B�K�C�h���C���͑傫���uQ�i�i���j�v�uS�i���S���j�v�uE�i�L�����j�v�uM�i�����̈�j�v��4�J�e�S���[�ɕ�����Ă���B
���̒��ŁuQ�V���[�Y�v�́A���i�̌���ѐ��܂��A��т��č����i�����ێ����邽�߂̊���߂����̂ł���B�����E�J���i�K���珳�F�\���A����ɂ͎s�̌�̃��C�t�T�C�N���S�̂ɂ�����CMC�i���w�E�����E�i���Ǘ��j�̗v����ԗ����Ă���B
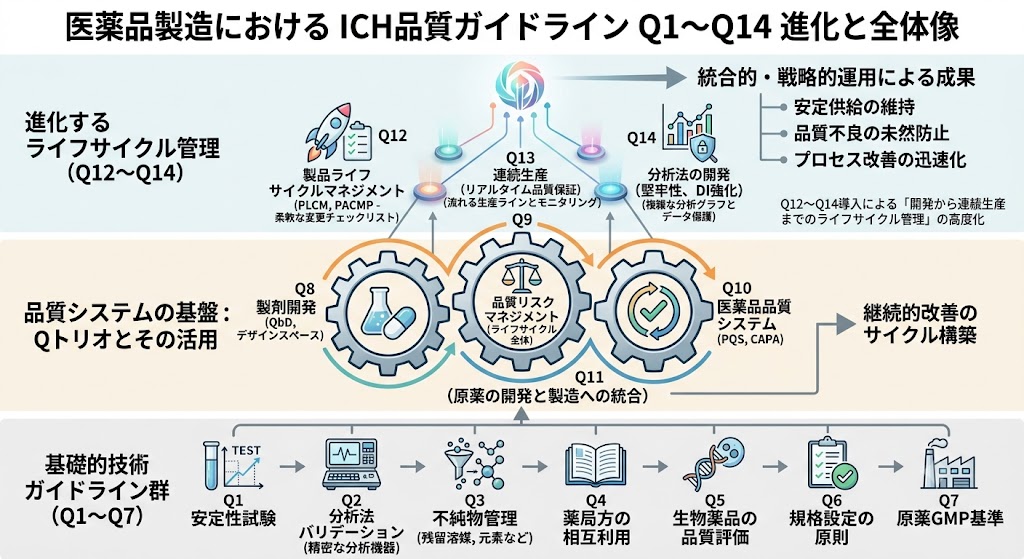
ICH Q1�`Q14�̊T�v
Q�V���[�Y�́A��b�I�ȕi�������̗v������ŐV�̐����Ǘ��V�X�e���܂ő���ɂ킽��B�e�K�C�h���C���̎��͈ȉ��̒ʂ�ł���B
- Q1�i���萫�����j�F���i�����x�⎼�x�A���Ȃǂ̊��v�������e����]�����A�L�����Ԃ⒙�@��ݒ肷�邽�߂̊�ł���B
- Q2�i���͖@�o���f�[�V�����j�F�i�����������镪�͎�@���A�ړI�Ƃ��鑪��ɑ��Đ��m���Ó��ł��邱�Ƃ��Ȋw�I�ɏؖ����邽�߂̗v���ł���B
- Q3�i�s�����j�F����ѐ��ܒ��ɑ��݂���c���n�}�⌳�f�s�����Ȃǂ̌��x�l�A��臒l��ݒ肵�A���S����S�ۂ���B
- Q4�i��Ǖ��j�F���ĉ��̖�Ǖ��ɂ����鎎�����@�┻���̍��ۓI�Ȓ��a��}����̂ł���B
- Q5�i�o�C�I���i�̕i���j�F�זE�|�{�ȂǂŐ��������o�C�I�e�N�m���W�[���p���i���L�̕i���]���A�E�C���X���S���Ȃǂ��K�肷��B
- Q6�i�K�i�y�ю������@�j�F�V��̏��F�\���ɂ����āA�ݒ肷�ׂ��i���K�i�i�������ڂ⍇�i��j�̍l�����������Ă���B
- Q7�i����GMP�j�F�L�������ł��錴��̐����Ǘ�����ѕi���Ǘ��̊�iGMP�j�����ۓI�ɓ��ꂵ�����̂ł���B
- Q8�i���܊J���j�FQbD�iQuality by Design�F�f�U�C���ɂ��i���j�̊T�O�����A���i�ƃv���Z�X�̐[���Ȋw�I�����Ɋ�Â��J���𐄏����Ă���B
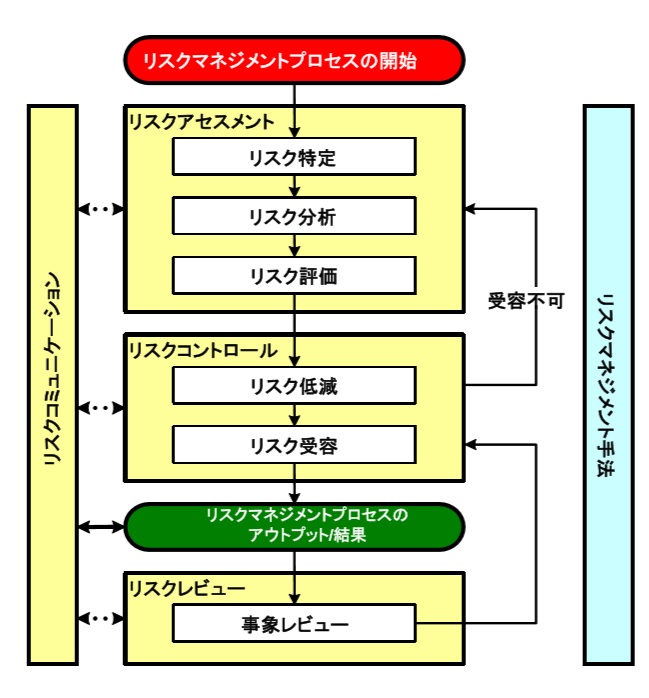
- Q9�i�i�����X�N�}�l�W�����g�j�F���i�̃��C�t�T�C�N���S�̂�ʂ��āA�i���Ɋւ��郊�X�N���Ȋw�I�ɕ]���A�Ǘ��A�`�B���邽�߂̑̌n�I�ȃA�v���[�`�����B
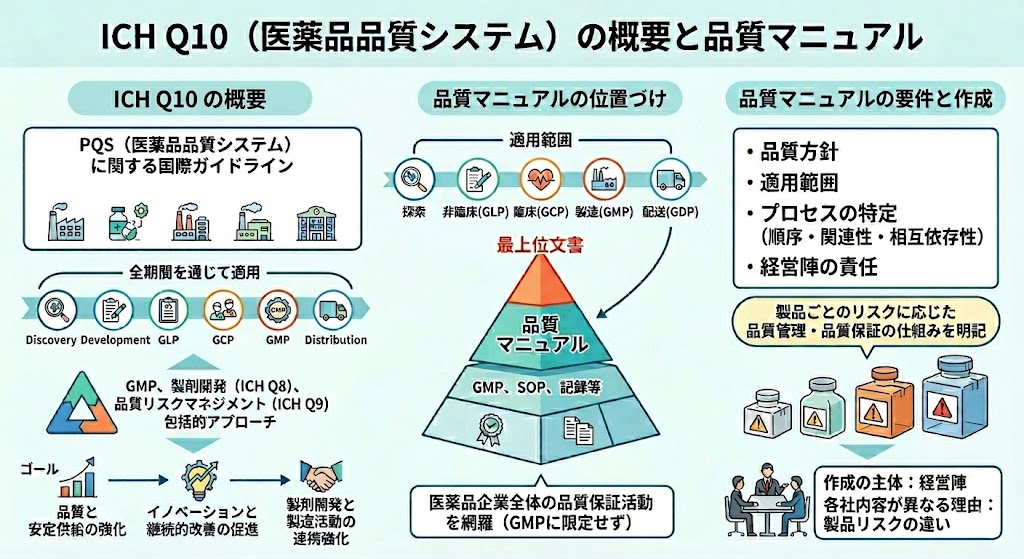
- Q10�i���i�i���V�X�e���j�F��Ƃ̌o�c�w���哱����p���I�ȕi�����P�ƃV�X�e���Ǘ��̃��f���iPQS�j�������B
- Q11�i����̊J���Ɛ����j�FQ8�̊T�O�������Ɋg�����A�o�������̑I�肩��Ǘ��헪�̍\�z�܂ł��K�肷��B
- Q12�i���C�t�T�C�N���}�l�W�����g�j�F���F�擾��̐������@�Ȃǂ̕ύX���A���X�N�x�[�X�ł��_������I�ɊǗ����邽�߂̘g�g�݂ł���B
- Q13�i�A�����Y�j�F�]���̃o�b�`���Y�ɑ���v�V�I�Ȑ�����@�u�A�����Y�v�̓����ƁA���̕i���ۏ̍l��������Ă���B
- Q14�i���͖@�̊J���j�FQ2��⊮���A���͖@�̊J���v���Z�X��QbD�̃A�v���[�`�����邱�ƂŁA���C�t�T�C�N����ʂ������͖@�̌��S�������߂�B
���i�����ɂ�����Q�V���[�Y�̏d�v��
����̕i���ۏɂ����āAQ8�AQ9�AQ10�́uICH Q�g���I�v�ƌĂ�A�]���̍ŏI�����d������v���Z�X�v�d���ւƃp���_�C���V�t�g�������炵���B�����Q11�A�����ĕύX�Ǘ��̍œK����ڎw��Q12������邱�ƂŁA�Ȋw�I�����ƃ��X�N�]���Ɋ�Â����_��ȕi���Ǘ��̊�Ղ��m������Ă���B
����ɁAQ13�i�A�����Y�j��Q14�i���͖@�J���j�́A������̃e�N�m���W�[�����ۂ̐����E���͌���֎������邽�߂̎w�j�ł���A����ƊE�S�̂őΉ����}���ƂȂ��Ă���B���i�����̍őO���ɂ����āA�����Q�V���[�Y�𐳂����������A���Ђ�PQS�i���i�i���V�X�e���j�֓I�m�ɗ��Ƃ����ނ��Ƃ́A���҂֎����I�����S�Ɉ��i�������������邽�߂̎g���ł���B
�i���Ɋւ���K�C�h���C���̎�ȓ��e
�@�����J���Ȃ���ʒm����A�����K���Ƃ��ē����ς݁i�X�e�b�v5�j�ƂȂ��Ă����ȃK�C�h���C���i2026�N���_�j���ȉ��Ɏ����B
| �K�C�h���C�� | �K�C�h���C���̎�ȓ��e | �ʒm���E �֘A�����N |
|---|---|---|
Q1 |
�y���萫�z ��Q1A�iR2�j�u���萫�����K�C�h���C���v�� ��Q1B�u�V����y�ѐV���܂̌����萫�����K�C�h���C���v�� ��Q1C�u�V���^�o�H���i���̈��萫�������т̎戵���Ɋւ���K�C�h���C���v�� ��Q1D�u����y�ѐ��܂̈��萫�����ւ̃u���P�b�e�B���O�@�y�у}�g���L�V���O�@�̓K�p�v�� ��Q1E�u���萫�f�[�^�̕]���Ɋւ���K�C�h���C���v�� ��Q1F�u�C����III�y��IV�ɂ����鏳�F�\���̂��߂̈��萫�������тɊւ���K�C�h���C���v�̔p�~�� |
2003.6.3 1997.5.28 1997.5.28 2002.7.31 2003.6.3 2006.7.3
Q1F�p�~�ʒm |
Q2 |
�y���͖@�o���f�[�V�����z ��Q2�iR1�j�u���͖@�o���f�[�V�����Ɋւ���e�L�X�g�i���{���ځj�v�� ��Q2�iR1�j�u���͖@�o���f�[�V�����Ɋւ���e�L�X�g�i���{���@�j�v�� |
1995.7.20 1997.10.28
���{���@���� |
Q3 |
�y�s�����z �@��Q3A�iR2�j�u�V�L�������ܗL���i�̂�������̕s�����Ɋւ���K�C�h���C���v�� �B��Q3B�iR2�j�u�V�L�������ܗL���i�̂������܂̕s�����Ɋւ���K�C�h���C���v�� �D��Q3C�iR3�`R6�j�u���i�̎c���n�}�K�C�h���C���v�y�ъe������ �I��Q3D�u���i�̌��f�s�����K�C�h���C���ɂ��āv�� |
2002.12.16 2003.6.24 1998.3.30 2015.9.30
�I���� |
Q4 |
�y��Ǖ��z ��ICH Q4B �K�C�h���C���i��Ǖ��e�L�X�g��ICH�n��ɂ����đ��ݗ��p���邽�߂̕]���y�ъ����j�� ���eAnnex�i�����ʕt�������j�� |
2009.5.26 2009.5.26
Annex 1���� �i����Annex��PMDA�����Q�Ɓj
|
Q5 |
�y������i�̕i���z ��ICH Q5A�iR1�j�u�E�C���X���S���]���v�� ��ICH Q5B�u��`�q�����\���̂̕��́v�� ��ICH Q5C�u������i�̈��萫�����v�� ��ICH Q5D�u�זE��܂̗R���A�����y�ѓ�����́v�� ��ICH Q5E�u�����H���̕ύX�ɂƂ��Ȃ��������^�������]���v�� |
2000.2.22 1998.1.6 1998.1.6 2000.7.14 2005.4.26
Q5E���� |
Q6 |
�y�K�i����ю������@�z ��ICH Q6A�u�V���i�̋K�i�y�ю������@�̐ݒ�v�� ��ICH Q6B�u������i�̋K�i�y�ю������@�̐ݒ�v�� |
2001.5.1 2001.5.1
Q6B���� |
Q7 |
�y����GMP�̃K�C�h���C���z �E����i���i�̗L�������j�Ɋւ��鐻���Ǘ��y�ѕi���Ǘ��iGMP�j�̍��ۓI�ȕW����������Ă���B |
2001.11.2 2016.3.8
Q&A���� |
Q8 (R2) |
�y���܊J���z �E���i����ѐ����H���̊J���ɑ���A�Ȋw�ƃ��X�N�Ɋ�Â��A�v���[�`�iQuality by Design�FQbD�j���L�q���Ă���B |
2010.6.28 ���� |
Q9 |
�y�i�����X�N�}�l�W�����g�z �E���i���C�t�T�C�N���S�̂�ʂ����A�i���ɑ��郊�X�N�̃A�Z�X�����g�A�R���g���[���A�R�~���j�P�[�V�����A����у��r���[�̌n���I�v���Z�X���L�q���Ă���B
|
2006.9.1 ���� |
Q10 |
�y���i�i���V�X�e���iPQS�j�z �E�K�ȕi��������L���鐻�i���P��I�ɋ������邽�߂̃V�X�e���m���E�ێ��ɂ��ċL�q���Ă���B |
2010.2.19 ���� |
Q11 |
�y����̊J���Ɛ����i���w�����^�����N���R�������j�z �EICH Q8�AQ9�AQ10�̌����ƊT�O�iQ�g���I�j���A����̊J���Ɛ����Ɋ֘A�Â��Đ������Ă���B |
2014.7.10 ���� |
Q12 |
�y���i���C�t�T�C�N���}�l�W�����g�iPLCM�j�z �E���i�̃��C�t�T�C�N���}�l�W�����g�ɂ�����Z�p��y�ыK����̍l�����Ɋւ���K�C�h���C���B |
2021.10.29 �֘A��� |
Q13 |
�y����y�ѐ��܂̘A�����Y�z �E����ѐ��܂̘A�����Y�̊J���A�����A����у��C�t�T�C�N���}�l�W�����g�Ɋւ���Ȋw�I�E�K����̍l�����������Ă���B |
2023.5.31 �֘A��� |
Q14 |
�y���͖@�̊J���z �E���͖@�J���ɂ�����Ȋw�I�A�v���[�`�i���͖@���C�t�T�C�N���}�l�W�����g�̊T�O�Ȃǁj�������A��茘�S�ȕ��͖@�̊J���𑣐i����B |
2025.10.9 �֘A��� |
�y�|�C���g�z
1. Q12�`Q14�����ɂ��u�J������A�����Y�܂ł̃��C�t�T�C�N���Ǘ��v�̍��x��
�ߔN���o���ꂽQ12�i���i���C�t�T�C�N���}�l�W�����g�j�AQ13�i�A�����Y�j�AQ14�i���͖@�̊J���j�ɂ��A���i�̕i���Ǘ��͐V���ȃX�e�[�W�ɓ˓����Ă���B����Q12��PACMP�i���F��ύX�Ǘ����{�v�揑�j�����p���邱�ƂŁA���O�̃��X�N�]���Ɋ�Â����_��ȕύX�Ǘ����\�ƂȂ�A�t�ܒ�����[�U�Ƃ�������������̃v���Z�X���P��v���ɐi�߂₷���Ȃ����B�܂��AQ13�������A�����Y��AQ14�����߂錘�S�ȕ��͖@�́A���A���^�C���ł̕i���ۏ�O��Ƃ��Ă��邽�߁A�@��̃I�[�f�B�b�g�g���C�����܂߂���茵�i�ȃf�[�^�C���e�O���e�B�iDI�j�Ή������߂���B
2. Q�g���I�iQ8, Q9, Q10�j���甭�W�����p���I���P�̃T�C�N���\�z
Q12�ȍ~�̐V�����K�C�h���C���Q�́A������Q8�i���܊J���j�AQ9�i�i�����X�N�}�l�W�����g�j�AQ10�i���i�i���V�X�e���j�Ƃ������uQ�g���I�v�̊�Ղ̏�ɐ��藧���Ă���B�K�C�h���C����P�Ȃ�K���Ƃ��đ�����̂ł͂Ȃ��A�������I���헪�I�ɉ^�p���A��E�������ɂ͍��{���������Ɋ�Â�CAPA�i�����[�u�E�\�h�[�u�j���m���ɋ@�\�����邱�Ƃ��d�v�ł���B���̈�A�̃V�X�e�������ꃌ�x���Œ蒅�����邱�Ƃ��A���苟���̈ێ���i���s�ǂ̖��R�h�~�ɒ�������B
�@
PMDA�u�i���Ɋւ���K�C�h���C���v�ŐV�̌�����������